METHOD 108 -
DETERMINATION OF PARTICULATE AND GASEOUS ARSENIC EMISSIONS
NOTE: This method does not include all of the
specifications (e.g.,
equipment and supplies) and procedures (e.g., sampling and analytical) essential to its
performance. Some material is incorporated by reference from other methods in
Appendix A to 40 CFR Part 60. Therefore, to obtain reliable results, persons
using this method should have a thorough knowledge of at least the following
additional test methods: Method 1, Method 2, Method 3, Method 5, and Method 12.
8.0 Sample Collection,
Preservation, Transport, and Storage.
10.0 Calibration and
Standardization.
10.2 Preparation of
Standard Solutions.
10.4 Spectrophotometer
Calibration Quality Control.
11.3 Spectrophotometer
Preparation.
11.5 Check for matrix
effects on the arsenic results.
12.0 Data Analysis and
Calculations.
14.0 Pollution
Prevention. [Reserved]
15.0 Waste Management.
[Reserved]
17.0 Tables, Diagrams,
Flowcharts, and Validation Data.
1.0 Scope and Application.
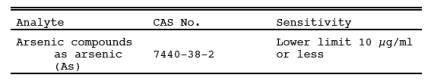
1.1 Analytes.
1.2 Applicability.
This method is
applicable for the determination of inorganic As emissions from stationary
sources as specified in an applicable subpart of the regulations.
1.3 Data Quality Objectives.
Adherence to the
requirements of this method will enhance the quality of the data obtained from
air pollutant sampling methods.
2.0 Summary of Method.
Particulate and
gaseous As emissions are withdrawn isokinetically from the source and are
collected on a glass mat filter and in water. The collected arsenic is then
analyzed by means of atomic absorption spectrophotometry (AAS).
3.0 Definitions. [Reserved]
4.0 Interferences.
Analysis for As by
flame AAS is sensitive to the chemical composition and to the physical
properties (e.g., viscosity,
pH) of the sample. The analytical procedure includes a check for matrix effects
(Section 11.5).
5.0 Safety.
5.1 This method may
involve hazardous materials, operations, and equipment. This test method may
not address all of the safety problems associated with its use. It is the
responsibility of the user to establish appropriate safety and health practices
and determine the applicability of regulatory limitations prior to performing
this test method.
5.2 Corrosive
reagents. The following reagents are hazardous. Personal protective equipment
and safe procedures that prevent chemical splashes are recommended. If contact
occurs, immediately flush with copious amounts of water for at least 15
minutes. Remove clothing under shower and decontaminate. Treat residual
chemical burns as thermal burns.
5.2.1 Hydrochloric
Acid (HCl). Highly corrosive liquid with toxic vapors. Vapors are highly irritating
to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis,
pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent
can be lethal to humans in a few minutes. Provide ventilation to limit
exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Hydrogen
Peroxide (H2O2). Very harmful to
eyes. 30% H2O2 can burn skin, nose,
and lungs.
5.2.3 Nitric Acid
(HNO3). Highly corrosive to eyes, skin, nose, and
lungs. Vapors are highly toxic and can cause bronchitis, pneumonia, or edema of
lungs. Reaction to inhalation may be delayed as long as 30 hours and still be
fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous
reaction may occur with organic materials such as solvents.
5.2.4 Sodium Hydroxide
(NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to
nose, throat, and lungs. Reacts exothermically with small amounts of water.
6.0 Equipment and Supplies.
6.1 Sample Collection.
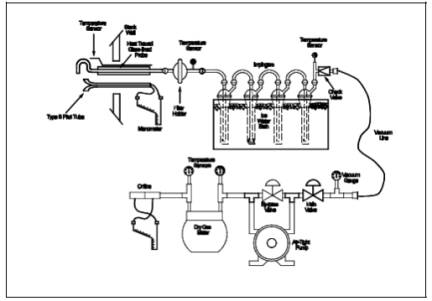
A schematic of the
sampling train used in performing this method is shown in Figure
108-1; it is similar to the Method 5 sampling train of 40 CFR Part 60,
Appendix A. The following items are required for sample collection:
6.1.1 Probe Nozzle,
Probe Liner, Pitot Tube, Differential Pressure Gauge, Filter Holder, Filter
Heating System, Temperature Sensor, Metering System, Barometer, and Gas Density
Determination Equipment. Same as Method 5,
Sections 6.1.1.1 to 6.1.1.7, 6.1.1.9, 6.1.2, and 6.1.3, respectively.
6.1.2 Impingers. Four
impingers connected in series with leak-free ground-glass fittings or any
similar leak-free noncontaminating fittings. For the first, third, and fourth
impingers, use the Greenburg-Smith design, modified by replacing the tip with a
1.3-cm ID (0.5-in.) glass tube extending to about 1.3 cm (0.5 in.) from the
bottom of the flask. For the second impinger, use the Greenburg-Smith design
with the standard tip. Modifications (e.g., flexible connections between the impingers, materials other than
glass, or flexible vacuum lines to connect the filter holder to the condenser)
are subject to the approval of the Administrator.
6.1.3 Temperature
Sensor. Place a temperature sensor, capable of measuring temperature to within
1 ºC (2 ºF), at the outlet of the fourth impinger for monitoring purposes.
6.2 Sample Recovery.
The following items
are required for sample recovery:
6.2.1 Probe-Liner and
Probe-Nozzle Brushes, Petri Dishes, Graduated Cylinder and/or Balance, Plastic
Storage Containers, and Funnel and Rubber Policeman. Same as Method 5, Sections 6.2.1 and 6.2.4 to 6.2.8,
respectively.
6.2.2 Wash Bottles.
Polyethylene (2).
6.2.3 Sample Storage
Containers. Chemically resistant, polyethylene or polypropylene for glassware
washes, 500- or 1000-ml.
6.3 Analysis.
The following items
are required for analysis:
6.3.1
Spectrophotometer. Equipped with an electrodeless discharge lamp and a
background corrector to measure absorbance at 193.7 nanometers (nm). For
measuring samples having less than 10 µg As/ml, use a vapor generator accessory
or a graphite furnace.
6.3.2 Recorder. To
match the output of the spectrophotometer.
6.3.3 Beakers. 150
ml.
6.3.4 Volumetric
Flasks. Glass 50-, 100-, 200-, 500-, and 1000-ml; and polypropylene, 50-ml.
6.3.5 Balance. To
measure within 0.5 g.
6.3.6 Volumetric
Pipets. 1-, 2-, 3-, 5-, 8-, and 10-ml.
6.3.7 Oven.
6.3.8 Hot Plate.
7.0 Reagents and Standards.
Unless otherwise
indicated, it is intended that all reagents conform to the specifications
established by the Committee on Analytical Reagents of the American Chemical
Society, where such specifications are available; otherwise, use the best
available grade.
7.1 Sample Collection
The following
reagents are required for sample collection:
7.1.1 Filters. Same
as Method 5, Section 7.1.1, except that the
filters need not be unreactive to SO2.
7.1.2 Silica Gel,
Crushed Ice, and Stopcock Grease. Same as Method 5, Sections 7.1.2, 7.1.4, and
7.1.5, respectively.
7.1.3 Water.
Deionized distilled to meet ASTM D 1193-77 or 91 (incorporated by reference)see § 61.18), Type 3. When high concentrations of organic matter
are not expected to be present, the KMnO4 test for
oxidizable organic matter may be omitted.
7.2 Sample Recovery.
7.2.1 0.1 N NaOH. Dissolve
4.00 g of NaOH in about 500 ml of water in a 1-liter volumetric flask. Then,
dilute to exactly 1.0 liter with water.
7.3 Analysis.
The following
reagents and standards are required for analysis:
7.3.1 Water. Same as
Section 7.1.3.
7.3.2 Sodium Hydroxide,
0.1 N. Same as in Section 7.2.1.
7.3.3 Sodium
Borohydride (NaBH4), 5 Percent Weight by Volume (W/V). Dissolve
50.0 g of NaBH4 in about 500 ml of 0.1 N NaOH in a 1-liter
volumetric flask. Then, dilute to exactly 1.0 liter with 0.1 N NaOH.
7.3.4 Hydrochloric
Acid, Concentrated.
7.3.5 Potassium
Iodide (KI), 30 Percent (W/V). Dissolve 300 g of KI in 500 ml of water in a 1
liter volumetric flask. Then, dilute to exactly 1.0 liter with water.
7.3.6 Nitric Acid,
Concentrated.
7.3.7 Nitric Acid,
0.8 N. Dilute 52 ml of concentrated HNO3 to
exactly 1.0 liter with water.
7.3.8 Nitric Acid, 50
Percent by Volume (V/V). Add 50 ml concentrated HNO3 to 50 ml water.
7.3.9 Stock Arsenic
Standard, 1 mg As/ml. Dissolve 1.3203 g of primary standard grade As2O3 in 20 ml of 0.1 N NaOH in a 150 ml beaker.
Slowly add 30 ml of concentrated HNO3. Heat the
resulting solution and evaporate just to dryness. Transfer the residue
quantitatively to a 1-liter volumetric flask, and dilute to 1.0 liter with
water.
7.3.10 Arsenic
Working Solution, 1.0 µg As/ml. Pipet exactly 1.0 ml of stock arsenic standard
into an acid-cleaned, appropriately labeled 1-liter volumetric flask containing
about 500 ml of water and 5 ml of concentrated HNO3. Dilute to exactly 1.0 liter with water.
7.3.11 Air. Suitable
quality for AAS analysis.
7.3.12 Acetylene.
Suitable quality for AAS analysis.
7.3.13 Nickel
Nitrate, 5 Percent Ni (W/V). Dissolve 24.780 g of nickel nitrate hexahydrate
[Ni(NO3)26H2O] in water in a 100-ml volumetric flask, and dilute to 100 ml with
water.
7.3.14 Nickel
Nitrate, 1 Percent Ni (W/V). Pipet 20 ml of 5 percent nickel nitrate solution
into a 100-ml volumetric flask, and dilute to exactly 100 ml with water.
7.3.15 Hydrogen
Peroxide, 3 Percent by Volume. Pipet 50 ml of 30 percent H2O2 into a 500-ml volumetric flask, and dilute to
exactly 500 ml with water.
7.3.16 Quality
Assurance Audit Samples. When making compliance determinations, and upon
availability, audit samples may be obtained from the appropriate EPA regional
Office or from the responsible enforcement authority.
NOTE: The responsible enforcement authority should be
notified at least 30 days prior to the test date to allow sufficient time for
sample delivery.
8.0 Sample Collection, Preservation, Transport, and Storage.
8.1 Pretest
Preparation. Follow the general procedure given in Method 5, Section 8.1, except the filter need
not be weighed, and the 200 ml of 0.1N NaOH and Container 4 should be tared to
within 0.5 g.
8.2 Preliminary
Determinations. Follow the general procedure given in Method 5, Section 8.2, except select the nozzle
size to maintain isokinetic sampling rates below 28 liters/min (1.0 cfm).
8.3 Preparation of
Sampling Train. Follow the general procedure given in Method 5, Section 8.3.
8.4 Leak-Check
Procedures. Same as Method 5, Section 8.4.
8.5 Sampling Train
Operation. Follow the general procedure given in Method
5, Section 8.5, except maintain isokinetic sampling flow rates below 28
liters/min (1.0 cfm). For each run, record the data required on a data sheet
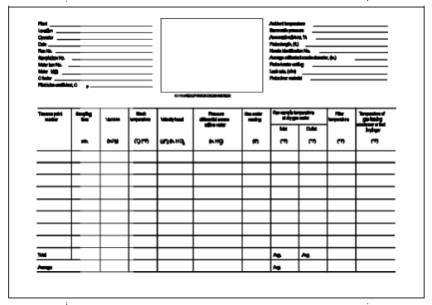
similar to the one shown in Figure 108-2.
8.6 Calculation of
Percent Isokinetic. Same as Method 5, Section 8.6.
8.7 Sample Recovery.
Same as Method 5, Section 8.7, except that
0.1 N NaOH is used as the cleanup solvent instead of acetone and that the
impinger water is treated as follows:
8.7.1 Container
Number 4 (Impinger Water). Clean each of the first three impingers and
connecting glassware in the following manner:
8.7.1.1 Wipe the
impinger ball joints free of silicone grease, and cap the joints.
8.7.1.2 Rotate and
agitate each of the first two impingers, using the impinger contents as a rinse
solution.
8.7.1.3 Transfer the
liquid from the first three impingers to Container Number 4. Remove the outlet
ball-joint cap, and drain the contents through this opening. Do not separate
the impinger parts (inner and outer tubes) while transferring their contents to
the container.
8.7.1.4 Weigh the
contents of Container No. 4 to within 0.5 g. Record in the log the weight of
liquid along with a notation of any color or film observed in the impinger
catch. The weight of liquid is needed along with the silica gel data to
calculate the stack gas moisture content.
NOTE: Measure and record the total amount of 0.1 N
NaOH used for rinsing under Sections 8.7.1.5 and 8.7.1.6.
8.7.1.5 Pour
approximately 30 ml of 0.1 NaOH into each of the first two impingers, and
agitate the impingers. Drain the 0.1 N NaOH through the outlet arm of each
impinger into Container Number 4. Repeat this operation a second time; inspect
the impingers for any abnormal conditions.
8.7.1.6 Wipe the ball
joints of the glassware connecting the impingers and the back half of the filter
holder free of silicone grease, and rinse each piece of glassware twice with
0.1 N NaOH; transfer this rinse into Container Number 4. (DO NOT RINSE or brush
the glass-fritted filter support.) Mark the height of the fluid level to
determine whether leakage occurs during transport. Label the container to
identify clearly its contents.
8.8 Blanks.
8.8.1 Sodium
Hydroxide. Save a portion of the 0.1 N NaOH used for cleanup as a blank. Take
200 ml of this solution directly from the wash bottle being used and place it
in a plastic sample container labeled "NaOH blank."
8.8.2 Water. Save a
sample of the water, and place it in a container labeled "H2O blank."
8.8.3 Filter. Save
two filters from each lot of filters used in sampling. Place these filters in a
container labeled "filter blank."
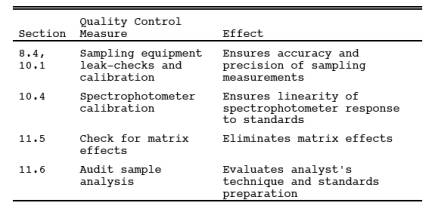
9.0 Quality Control.
9.1 Miscellaneous
Quality Control Measures.
9.2 Volume Metering
System Checks. Same as Method 5, Section 9.2.
10.0 Calibration and Standardization.
NOTE: Maintain a laboratory log of all calibrations.
10.1 Sampling Equipment.
Same as Method 5, Section 10.0.
10.2 Preparation of Standard Solutions.
10.2.1 For the high
level procedure, pipet 1, 3, 5, 8, and 10 ml of the 1.0 mg As/ml stock solution
into separate 100 ml volumetric flasks, each containing 5 ml of concentrated
HNO3. Dilute to the mark with water.
10.2.2 For the low
level vapor generator procedure, pipet 1, 2, 3, and 5 ml of 1.0 µg As/ml
standard solution into separate reaction tubes. Dilute to the mark with water.
10.2.3 For the low
level graphite furnace procedure,pipet 1, 5, 10 and 15 ml of 1.0 µg As/ml
standard solution into separate flasks along with 2 ml of the 5 percent nickel
nitrate solution and 10 ml of the 3 percent H202 solution. Dilute to the mark with water.
10.3 Calibration Curve.
Analyze a 0.8 N HNO3 blank and each standard solution according to the procedures
outlined in Section 11.4.1. Repeat this procedure on each standard solution
until two consecutive peaks agree within 3 percent of their average value.
Subtract the average peak height (or peak area) of the blank - which must be
less than 2 percent of recorder full scale - from the averaged peak height of
each standard solution. If the blank absorbance is greater than 2 percent of
full-scale, the probable cause is As contamination of a reagent or carry-over
of As from a previous sample. Prepare the calibration curve by plotting the corrected
peak height of each standard solution versus the corresponding final total As
weight in the solution.
10.4 Spectrophotometer Calibration Quality Control.
Calculate the least
squares slope of the calibration curve.
The line must pass through the origin or through a point no further from
the origin than ±2 percent of the recorder full scale. Multiply the corrected
peak height by the reciprocal of the least squares slope to determine the
distance each calibration point lies from the theoretical calibration line. The
difference between the calculated concentration values and the actual
concentrations (e.g., 1, 3,
5, 8, and 10 mg As for the high-level procedure) must be less than 7 percent
for all standards.
NOTE: For instruments equipped with direct concentration
readout devices, preparation of a standard curve will not be necessary. In all
cases, follow calibration and operational procedures in the manufacturers'
instruction manual.
11.0 Analytical Procedure.
11.1 Sample Loss Check.
Prior to analysis,
check the liquid level in Containers Number 2 and Number 4. Note on the
analytical data sheet whether leakage occurred during transport. If a
noticeable amount of leakage occurred, either void the sample or take steps,
subject to the approval of the Administrator, to adjust the final results.
11.2 Sample Preparation.
11.2.1 Container
Number 1 (Filter). Place the filter and loose particulate matter in a 150 ml
beaker. Also, add the filtered solid material from Container Number 2 (see
Section 11.2.2). Add 50 ml of 0.1 N NaOH. Then stir and warm on a hot plate at
low heat (do not boil) for about 15 minutes. Add 10 ml of concentrated HNO3, bring to a boil, then simmer for about 15 minutes. Filter the
solution through a glass fiber filter. Wash with hot water, and catch the
filtrate in a clean 150 ml beaker. Boil the filtrate, and evaporate to dryness.
Cool, add 5 ml of 50 percent HNO3, and then warm and
stir. Allow to cool. Transfer to a 50-ml volumetric flask, dilute to volume
with water, and mix well.
11.2.2 Container
Number 2 (Probe Wash).
11.2.2.1 Filter
(using a glass fiber filter) the contents of Container Number 2 into a 200 ml
volumetric flask. Combine the filtered (solid) material with the contents of
Container Number 1 (Filter).
11.2.2.2 Dilute the
filtrate to exactly 200 ml with water. Then pipet 50 ml into a 150 ml beaker.
Add 10 ml of concentrated HNO3, bring to a boil, and
evaporate to dryness. Allow to cool, add 5 ml of 50 percent HNO3, and then warm and stir. Allow the solution to cool, transfer to a
50-ml volumetric flask, dilute to volume with water, and mix well.
11.2.3 Container
Number 4 (Impinger Solution). Transfer the contents of Container Number 4 to a
500 ml volumetric flask, and dilute to exactly 500-ml with water. Pipet 50 ml
of the solution into a 150-ml beaker. Add 10 ml of concentrated HNO3, bring to a boil, and evaporate to dryness. Allow to cool, add 5
ml of 50 percent HNO3, and then warm and stir. Allow the solution to
cool, transfer to a 50-ml volumetric flask, dilute to volume with water, and
mix well.
11.2.4 Filter Blank.
Cut each filter into strips, and treat each filter individually as directed in
Section 11.2.1, beginning with the sentence, "Add 50 ml of 0.1 N
NaOH."
11.2.5 Sodium
Hydroxide and Water Blanks. Treat separately 50 ml of 0.1 N NaOH and 50 ml
water, as directed under Section 11.2.3, beginning with the sentence,
"Pipet 50 ml of the solution into a 150-ml beaker."
11.3 Spectrophotometer Preparation.
Turn on the power;
set the wavelength, slit width, and lamp current. Adjust the background
corrector as instructed by the manufacturer's manual for the particular atomic
absorption spectrophotometer. Adjust the burner and flame characteristics as
necessary.
11.4 Analysis.
Calibrate the analytical
equipment and develop a calibration curve as outlined in Sections 10.2 through
10.4
11.4.1 Arsenic
Samples. Analyze an appropriately sized aliquot of each diluted sample (from
Sections 11.2.1 through 11.2.3) until two consecutive peak heights agree within
3 percent of their average value. If applicable, follow the procedures outlined
in Section 11.4.1.1. If the sample concentration falls outside the range of the
calibration curve, make an appropriate dilution with 0.8 N HNO3 so that the final concentration falls within the range of the
curve. Using the calibration curve, determine the arsenic concentration in each
sample fraction.
NOTE: Because instruments vary between
manufacturers, no detailed operating instructions will be given here. Instead,
the instrument manufacturer's detailed operating instructions should be
followed.
11.4.1.1 Arsenic
Determination at Low Concentration. The lower limit of flame AAS is 10 µg
As/ml. If the arsenic concentration of any sample is at a lower level, use the
graphite furnace or vapor generator which is available as an accessory
component. Flame, graphite furnace, or vapor generators may be used for samples
whose concentrations are between 10 and 30 µg/ml. Follow the manufacturer's
instructions in the use of such equipment.
11.4.1.1.1 Vapor
Generator Procedure. Place a sample containing between 0 and 5 µg of arsenic in
the reaction tube, and dilute to 15 ml with water. Since there is some trial and
error involved in this procedure, it may be necessary to screen the samples by
conventional atomic absorption until an approximate concentration is
determined. After determining the approximate concentration, adjust the volume
of the sample accordingly. Pipet 15 ml of concentrated HCl into each tube. Add
1 ml of 30 percent KI solution. Place the reaction tube into a 50 ºC (120 ºF)
water bath for 5 minutes. Cool to room temperature. Connect the reaction tube
to the vapor generator assembly. When the instrument response has returned to
baseline, inject 5.0 ml of 5 percent NaBH4, and
integrate the resulting spectrophotometer signal over a 30-second time period.
11.4.1.1.2 Graphite
Furnace Procedure. Dilute the digested sample so that a 5 ml aliquot contains
less than 1.5 µg of arsenic. Pipet 5 ml of this digested solution into a 10-ml
volumetric flask. Add 1 ml of the 1 percent nickel nitrate solution, 0.5 ml of
50 percent HNO3, and 1 ml of the 3 percent hydrogen peroxide
and dilute to 10 ml with water. The sample is now ready for analysis.
11.4.1.2 Run a blank
(0.8 N HNO3) and standard at least after every five samples
to check the spectrophotometer calibration. The peak height of the blank must
pass through a point no further from the origin than ±2 percent of the recorder
full scale. The difference between the measured concentration of the standard
(the product of the corrected average peak height and the reciprocal of the
least squares slope) and the actual concentration of the standard must be less
than 7 percent, or recalibration of the analyzer is required.
11.4.1.3 Determine
the arsenic concentration in the filter blank (i.e., the average of the two blank values from each
lot).
11.4.2 Container
Number 3 (Silica Gel). This step may be conducted in the field. Weigh the spent
silica gel (or silica gel plus impinger) to the nearest 0.5 g; record this
weight.
11.5 Check for matrix effects on the arsenic results.
Same as Method 12, Section 11.5.
11.6 Audit Sample Analysis.
11.6.1 When the
method is used to analyze samples to demonstrate compliance with a source
emission regulation, a set of EPA audit samples must be analyzed, subject to
availability.
11.6.2 Concurrently
analyze the audit samples and the compliance samples in the same manner to
evaluate the technique of the analyst and the standards preparation.
NOTE: It is recommended that known quality control
samples be analyzed prior to the compliance and audit sample analyses to
optimize the system accuracy and precision. These quality control samples may
be obtained by contacting the appropriate EPA regional Office or the
responsible enforcement authority.
11.6.3 The same
analyst, analytical reagents, and analytical system shall be used for the
compliance samples and the EPA audit samples. If this condition is met,
duplicate auditing of subsequent compliance analyses for the same enforcement
agency within a 30-day period is waived. An audit sample set may not be used to
validate different sets of compliance samples under the jurisdiction of
separate enforcement agencies, unless prior arrangements have been made with
both enforcement agencies.
11.7 Audit Sample Results.
11.7.1 Calculate the
audit sample concentrations in g/m3 and
submit results using the instructions provided with the audit samples.
11.7.2 Report the
results of the audit samples and the compliance determination samples along
with their identification numbers, and the analyst's name to the responsible
enforcement authority. Include this information with reports of any subsequent
compliance analyses for the same enforcement authority during the 30-day
period.
11.7.3 The
concentrations of the audit samples obtained by the analyst shall agree within 10
percent of the actual concentrations. If the 10 percent specification is not
met, reanalyze the compliance and audit samples, and include initial and
reanalysis values in the test report.
11.7.4 Failure to
meet the 10 percent specification may require retests until the audit problems
are resolved. However, if the audit results do not affect the compliance or
noncompliance status of the affected facility, the Administrator may waive the
reanalysis requirement, further audits, or retests and accept the results of
the compliance test. While steps are being taken to resolve audit analysis
problems, the Administrator may also choose to use the data to determine the
compliance or noncompliance status of the affected facility.
12.0 Data Analysis and Calculations.
12.1 Nomenclature.



12.2 Average Dry Gas
Meter Temperatures (Tm) and Average Orifice Pressure Drop (∆H). See
data sheet (Figure 108-2).
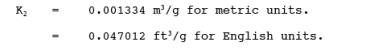
12.3 Dry Gas Volume.
Using data from this test, calculate Vm(std) according
to the procedures outlined in Method 5, Section
12.3.
12.4 Volume of Water
Vapor.
![]()
where:
12.5 Moisture
Content.

12.6 Amount of
Arsenic Collected.
12.6.1 Calculate the
amount of arsenic collected in each part of the sampling train, as follows:
![]()
12.6.2 Calculate the
total amount of arsenic collected in the sampling train as follows:

12.7 Calculate the arsenic
concentration in the stack gas (dry basis, adjusted to standard conditions) as
follows:
![]()
where:

12.8 Stack Gas
Velocity and Volumetric Flow Rate. Calculate the average stack gas velocity and
volumetric flow rate using data obtained in this method and the equations in Sections 12.2 and 12.3 of Method 2.
12.9 Pollutant Mass
Rate. Calculate the arsenic mass emission rate as follows:
![]()
12.10 Isokinetic
Variation. Same as Method 5, Section 12.11.
13.0 Method Performance.
13.1 Sensitivity. The
lower limit of flame AAS 10 µg As/ml. The analytical procedure includes provisions
for the use of a graphite furnace or vapor generator for samples with a lower
arsenic concentration.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
Same as References 1
through 9 of Method 5, Section 17.0, with the
addition of the following:
1. Perkin Elmer
Corporation. Analytical Methods for Atomic Absorption Spectrophotometry.
303-0152. Norwalk, Connecticut. September 1976. pp. 5-6.
2. Standard
Specification for Reagent Water. In: Annual Book of American Society for
Testing and Materials Standards. Part 31: Water, Atmospheric Analysis. American
Society for Testing and Materials. Philadelphia, PA. 1974. pp. 40-42.
3. Stack Sampling
Safety Manual (Draft). U.S. Environmental Protection Agency, Office of Air
Quality Planning and Standard, Research Triangle Park, NC. September 1978.
17.0 Tables, Diagrams, Flowcharts, and Validation Data.
Figure
108-1. Arsenic Sampling Train
Figure
108-2. Arsenic Field Data Sheet.