METHOD 320
MEASUREMENT OF VAPOR PHASE ORGANIC AND INORGANIC EMISSIONS BY EXTRACTIVE
FOURIER TRANSFORM INFRARED (FTIR) SPECTROSCOPY
Appendix A of part 63 is
amended by adding, in numerical order, Methods 320 and 321 to read as follows:
Appendix A to Part 63-Test Methods
1.2 Method Range and
Sensitivity.
2.3 Reference Spectra
Availability.
4.1.1 Background
Interference.
4.2 Sampling System
Interferences.
6.3 Sampling
Line/Heating System.
6.4 Gas Distribution
Manifold.
6.6 Calibration/Analyte
Spike Assembly.
6.9
Polytetrafluoroethane Tubing.
7.1 Analyte(s) and
Tracer Gas.
7.2 Calibration
Transfer Standard(s).
8.0 Sampling and
Analysis Procedure.
8.1 Pretest
Preparations and Evaluations.
8.1.4. Fractional
Reproducibility Uncertainty (FRUi).
8.1.6 Calculate the
Minimum Analyte Uncertainty, MAU
8.4 Data Storage
Requirements.
8.6.1 Calibration
Transfer Standard.
8.8 Sampling QA and
Reporting.
10.0 Calibration and
Standardization.
10.1 Signal-to-Noise
Ratio (S/N).
11.0 Data Analysis and
Calculations.
13.0 Method Validation
Procedure.
13.3 Simultaneous
Measurements With Two FTIR Systems.
2.0 APPLICABILITY AND
ANALYTICAL PRINCIPLE
3.0 GENERAL PRINCIPLES
OF PROTOCOL REQUIREMENTS
3.1 Verifiability and
Reproducibility of Results.
3.2 Transfer of
Reference Spectra.
3.3 Evaluation of FTIR
Analyses.
3.3.1
Sample-Independent Factors.
3.3.2 Sample-Dependent
Factors.
4.0 PRE-TEST
PREPARATIONS AND EVALUATIONS
4.1 Identify Test
Requirements.
4.2 Identify Potential
Interferants.
4.3 Select and Evaluate
the Sampling System.
4.4 Select
Spectroscopic System.
4.5 Select Calibration
Transfer Standards (CTS's).
4.6 Prepare Reference
Spectra.
4.7 Select Analytical
Regions.
4.8 Determine
Fractional Reproducibility Uncertainties.
4.9 Identify Known
Interferants.
4.10 Prepare
Computerized Analytical Programs.
4.11 Determine the
Fractional Calibration Uncertainty.
4.12 Verify System
Configuration Suitability.
5.0 SAMPLING AND
ANALYSIS PROCEDURE
5.1 Analysis System
Assembly and Leak-Test.
5.2 Verify Instrumental
Performance.
5.3 Determine the
Sample Absorption Pathlength.
5.5 Quantify Analyte
Concentrations.
5.6 Determine
Fractional Analysis Uncertainty.
6.1 Qualitatively
Confirm the Assumed Matrix.
6.2 Quantitatively
Evaluate Fractional Model Uncertainty (FMU).
6.3 Estimate Overall
Concentration Uncertainty (OCU).
APPENDIX A TO ADDENDUM TO METHOD 320
A.2 Definitions of
Mathematical Symbols.
APPENDIX B TO ADDENDUM
TO METHOD 320
APPENDIX C TO ADDENDUM
TO METHOD 320
APPENDIX D TO ADDENDUM
TO METHOD 320
APPENDIX E TO ADDENDUM
TO METHOD 320
APPENDIX F OF ADDENDUM
TO METHOD 320
APPENDIX G TO ADDENDUM
TO METHOD 320
APPENDIX H OF ADDENDUM
TO METHOD 320
APPENDIX I TO ADDENDUM
TO METHOD 320
APPENDIX J OF ADDENDUM
TO METHOD 320
1.0 Introduction.
Persons unfamiliar with
basic elements of FTIR spectroscopy should not attempt to use this method. This
method describes sampling and analytical procedures for extractive emission
measurements using Fourier transform infrared (FTIR) spectroscopy. Detailed
analytical procedures for interpreting infrared spectra are described in the
"Protocol for the Use of Extractive Fourier Transform Infrared (FTIR) Spectrometry
in Analyses of Gaseous Emissions from Stationary Sources," hereafter
referred to as the "Protocol." Definitions not given in this method
are given in appendix A of the Protocol. References to specific sections in the
Protocol are made throughout this Method. For additional information refer to
references 1 and 2, and other EPA reports, which describe the use of FTIR
spectrometry in specific field measurement applications and validation tests.
The sampling procedure described here is extractive. Flue gas is extracted
through a heated gas transport and handling system. For some sources, sample
conditioning systems may be applicable. Some examples are given in this method.
Note: sample conditioning systems may be used providing the method validation
requirements in Sections 9.2 and 13.0 of this method are met.
1.1 Scope and Applicability.
1.1.1 Analytes. Analytes
include hazardous air pollutants (HAPs) for which EPA reference spectra have
been developed. Other compounds can also be measured with this method if
reference spectra are prepared according to section 4.6 of the protocol.
1.1.2 Applicability. This
method applies to the analysis of vapor phase organic or inorganic compounds
which absorb energy in the mid-infrared spectral region, about 400 to 4000 cm-1 (25 to 2.5 µm). This method is used to determine compound-specific
concentrations in a multi-component vapor phase sample, which is contained in a
closed-path gas cell. Spectra of samples are collected using double beam
infrared absorption spectroscopy. A computer program is used to analyze spectra
and report compound concentrations.
1.2 Method Range and Sensitivity.
Analytical range and
sensitivity depend on the frequency-dependent analyte absorptivity, instrument
configuration, data collection parameters, and gas stream composition.
Instrument factors include: (a) spectral resolution, (b) interferometer signal
averaging time, (c) detector sensitivity and response, and (d) absorption path
length.
1.2.1 For any optical
configuration the analytical range is between the absorbance values of about
.01 (infrared transmittance relative to the background = 0.98) and 1.0 (T =
0.1). (For absorbance > 1.0 the relation between absorbance and
concentration may not be linear.)
1.2.2 The concentrations
associated with this absorbance range depend primarily on the cell path length
and the sample temperature. An analyte absorbance greater than 1.0, can be
lowered by decreasing the optical path length. Analyte absorbance increases
with a longer path length. Analyte detection also depends on the presence of
other species exhibiting absorbance in the same analytical region.
Additionally, the estimated lower absorbance (A) limit (A = 0.01) depends on
the root mean square deviation (RMSD) noise in the analytical region.
1.2.3 The concentration
range of this method is determined by the choice of optical configuration.
1.2.3.1 The absorbance for
a given concentration can be decreased by decreasing the path length or by
diluting the sample. There is no practical upper limit to the measurement
range.
1.2.3.2 The analyte
absorbance for a given concentration may be increased by increasing the cell
path length or (to some extent) using a higher resolution. Both modifications
also cause a corresponding increased absorbance for all compounds in the
sample, and a decrease in the signal throughput. For this reason the practical
lower detection range (quantitation limit) usually depends on sample
characteristics such as moisture content of the gas, the presence of other
interferants, and losses in the sampling system.
1.3 Sensitivity.
The limit of sensitivity
for an optical configuration and integration time is determined using appendix
D of the Protocol: Minimum Analyte Uncertainty, (MAU). The MAU depends on the
RMSD noise in an analytical region, and on the absorptivity of the analyte in
the same region.
1.4 Data Quality.
Data quality shall be
determined by executing Protocol pre-test procedures in appendices B to H of
the protocol and post-test procedures in appendices I and J of the protocol.
1.4.1 Measurement
objectives shall be established by the choice of detection limit (DLi) and analytical uncertainty (AUi) for each analyte.
1.4.2 An instrumental
configuration shall be selected. An estimate of gas composition shall be made
based on previous test data, data from a similar source or information gathered
in a pre-test site survey. Spectral interferants shall be identified using the
selected DLi and AUi and band areas from reference
spectra and interferant spectra. The baseline noise of the system shall be
measured in each analytical region to determine the MAU of the instrument
configuration for each analyte and interferant (MIUi).
1.4.3 Data quality for the
application shall be determined, in part, by measuring the RMS (root mean
square) noise level in each analytical spectral region (appendix C of the
Protocol). The RMS noise is defined as the RMSD of the absorbance values in an
analytical region from the mean absorbance value in the region.
1.4.4 The MAU is the
minimum analyte concentration for which the AUi can be
maintained; if the measured analyte concentration is less than MAUi, then data quality are unacceptable.
2.0 Summary of Method.
2.1 Principle.
References 4 through 7
provide background material on infrared spectroscopy and quantitative analysis.
A summary is given in this section.
2.1.1 Infrared absorption
spectroscopy is performed by directing an infrared beam through a sample to a
detector. The frequency-dependent infrared absorbance of the sample is measured
by comparing this detector signal (single beam spectrum) to a signal obtained
without a sample in the beam path (background).
2.1.2 Most molecules
absorb infrared radiation and the absorbance occurs in a characteristic and
reproducible pattern. The infrared spectrum measures fundamental molecular
properties and a compound can be identified from its infrared spectrum alone.
2.1.3 Within constraints,
there is a linear relationship between infrared absorption and compound
concentration. If this frequency dependent relationship (absorptivity) is known
(measured), it can be used to determine compound concentration in a sample
mixture.
2.1.4 Absorptivity is
measured by preparing, in the laboratory, standard samples of compounds at
known concentrations and measuring the FTIR "reference spectra" of
these standard samples. These "reference spectra" are then used in
sample analysis: (1) compounds are detected by matching sample absorbance bands
with bands in reference spectra, and (2) concentrations are measured by
comparing sample band intensities with reference band intensities.
2.1.5 This method is
self-validating provided that the results meet the performance requirement of
the QA spike in sections 8.6.2 and 9.0 of this method, and results from a
previous method validation study support the use of this method in the
application.
2.2 Sampling and Analysis.
In extractive sampling a
probe assembly and pump are used to extract gas from the exhaust of the
affected source and transport the sample to the FTIR gas cell. Typically, the
sampling apparatus is similar to that used for single-component continuous
emission monitor (CEM) measurements.
2.2.1 The digitized
infrared spectrum of the sample in the FTIR gas cell is measured and stored on
a computer. Absorbance band intensities in the spectrum are related to sample
concentrations by what is commonly referred to as Beer's Law.
![]()
where:

2.2.2 Analyte spiking is
used for quality assurance (QA). In this procedure (section 8.6.2 of this
method) an analyte is spiked into the gas stream at the back end of the sample
probe. Analyte concentrations in the spiked samples are compared to analyte
concentrations in unspiked samples. Since the concentration of the spike is
known, this procedure can be used to determine if the sampling system is
removing the spiked analyte(s) from the sample stream.
2.3 Reference Spectra Availability.
Reference spectra of over
100 HAPs are available in the EPA FTIR spectral library on the EMTIC (Emission
Measurement Technical Information Center) computer bulletin board service and
at internet address https://info.arnold.af.mil/epa/welcome.htm.
Reference spectra for HAPs, or other analytes, may also be prepared according
to section 4.6 of the Protocol.
2.4 Operator Requirements.
The FTIR analyst shall be
trained in setting up the instrumentation, verifying the instrument is
functioning properly, and performing routine maintenance. The analyst must
evaluate the initial sample spectra to determine if the sample matrix is
consistent with pre-test assumptions and if the instrument configuration is
suitable. The analyst must be able to modify the instrument configuration, if
necessary.
2.4.1 The spectral
analysis shall be supervised by someone familiar with EPA FTIR Protocol
procedures.
2.4.2 A technician trained
in instrumental test methods is qualified to install and operate the sampling
system. This includes installing the probe and heated line assembly, operating
the analyte spike system, and performing moisture and flow measurements.
3.0 Definitions.
See appendix A of the
Protocol for definitions relating to infrared spectroscopy. Additional
definitions are given in sections 3.1 through 3.29.
3.1 Analyte.
A compound that this
method is used to measure. The term "target analyte" is also used.
This method is multi-component and a number of analytes can be targeted for a
test.
3.2 Reference Spectrum.
Infrared spectrum of an
analyte prepared under controlled, documented, and reproducible laboratory
conditions according to procedures in section 4.6 of the Protocol. A library of
reference spectra is used to measure analytes in gas samples.
3.3 Standard Spectrum.
A spectrum that has been
prepared from a reference spectrum through a (documented) mathematical
operation. A common example is de-resolving of reference spectra to
lower-resolution standard spectra (Protocol, appendix K to the addendum of this
method). Standard spectra, prepared by approved, and documented, procedures can
be used as reference spectra for analysis.
3.4 Concentration.
In this method
concentration is expressed as a molar concentration, in ppm-meters, or in
(ppm-meters)/K, where K is the absolute temperature (Kelvin). The latter units
allow the direct comparison of concentrations from systems using different
optical configurations or sampling temperatures.
3.5 Interferant.
A compound in the sample
matrix whose infrared spectrum overlaps with part of an analyte spectrum. The
most accurate analyte measurements are achieved when reference spectra of
interferants are used in the quantitative analysis with the analyte reference
spectra. The presence of an interferant can increase the analytical uncertainty
in the measured analyte concentration.
3.6 Gas Cell.
A gas containment cell
that can be evacuated. It is equipped with the optical components to pass the
infrared beam through the sample to the detector. Important cell features
include: path length (or range if variable), temperature range, materials of
construction, and total gas volume.
3.7 Sampling System.
Equipment used to extract
the sample from the test location and transport the sample gas to the FTIR
analyzer. This includes sample conditioning systems.
3.8 Sample Analysis.
The process of interpreting
the infrared spectra to obtain sample analyte concentrations. This process is
usually automated using a software routine employing a classical least squares
(cls), partial least squares (pls), or K- or P- matrix method.
3.9 One hundred percent line.
A double beam
transmittance spectrum obtained by combining two background single beam
spectra. Ideally, this line is equal to 100 percent transmittance (or zero
absorbance) at every frequency in the spectrum. Practically, a zero absorbance
line is used to measure the baseline noise in the spectrum.
3.10 Background Deviation.
A deviation from 100
percent transmittance in any region of the 100 percent line. Deviations greater
than ± 5 percent in an analytical region are unacceptable (absorbance of 0.021
to -0.022). Such deviations indicate a change in the instrument throughput
relative to the background single beam.
3.11 Batch Sampling.
A procedure where spectra
of discreet, static samples are collected. The gas cell is filled with sample
and the cell is isolated. The spectrum is collected. Finally, the cell is
evacuated to prepare for the next sample.
3.12 Continuous Sampling.
A procedure where spectra
are collected while sample gas is flowing through the cell at a measured rate.
3.13 Sampling resolution.
The spectral resolution
used to collect sample spectra.
3.14 Truncation.
Limiting the number of
interferogram data points by deleting points farthest from the center burst
(zero path difference, ZPD).
3.15 Zero filling.
The addition of points to
the interferogram. The position of each added point is interpolated from
neighboring real data points. Zero filling adds no information to the
interferogram, but affects line shapes in the absorbance spectrum (and possibly
analytical results).
3.16 Reference CTS.
Calibration Transfer
Standard spectra that were collected with reference spectra.
3.17 CTS Standard.
CTS spectrum produced by
applying a deresolution procedure to a reference CTS.
3.18 Test CTS.
CTS spectra collected at
the sampling resolution using the same optical configuration as for sample
spectra. Test spectra help verify the resolution, temperature and path length
of the FTIR system.
3.19 RMSD.
Root Mean Square
Difference, defined in EPA FTIR Protocol, appendix A.
3.20 Sensitivity.
The noise-limited
compound-dependent detection limit for the FTIR system configuration. This is
estimated by the MAU. It depends on the RMSD in an analytical region of a zero
absorbance line.
3.21 Quantitation Limit.
The lower limit of
detection for the FTIR system configuration in the sample spectra. This is
estimated by mathematically subtracting scaled reference spectra of analytes
and interferences from sample spectra, then measuring the RMSD in an analytical
region of the subtracted spectrum. Since the noise in subtracted sample spectra
may be much greater than in a zero absorbance spectrum, the quantitation limit
is generally much higher than the sensitivity. Removing spectral interferences
from the sample or improving the spectral subtraction can lower the
quantitation limit toward (but not below) the sensitivity.
3.22 Independent Sample.
A unique volume of sample
gas; there is no mixing of gas between two consecutive independent samples. In
continuous sampling two independent samples are separated by at least 5 cell
volumes. The interval between independent measurements depends on the cell
volume and the sample flow rate (through the cell).
3.23 Measurement.
A single spectrum of flue
gas contained in the FTIR cell.
3.24 Run.
A run consists of a series
of measurements. At a minimum a run includes 8 independent measurements spaced
over 1 hour.
3.25 Validation.
Validation of FTIR
measurements is described in sections 13.0 through 13.4 of this method.
Validation is used to verify the test procedures for measuring specific
analytes at a source. Validation provides proof that the method works under
certain test conditions.
3.26 Validation Run.
A validation run consists
of at least 24 measurements of independent samples. Half of the samples are
spiked and half are not spiked. The length of the run is determined by the
interval between independent samples.
3.27 Screening. Screening
is used when there is
little or no available information about a source. The purpose of screening is
to determine what analytes are emitted and to obtain information about
important sample characteristics such as moisture, temperature, and
interferences. Screening results are semi-quantitative (estimated
concentrations) or qualitative (identification only). Various optical and
sampling configurations may be used. Sample conditioning systems may be
evaluated for their effectiveness in removing interferences. It is unnecessary
to perform a complete run under any set of sampling conditions. Spiking is not
necessary, but spiking can be a useful screening tool for evaluating the
sampling system, especially if a reactive or soluble analyte is used for the
spike.
3.28 Emissions Test.
An FTIR emissions test is
performed according specific sampling and analytical procedures. These procedures,
for the target analytes and the source, are based on previous screening and
validation results. Emission results are quantitative. A QA spike (sections
8.6.2 and 9.2 of this method) is performed under each set of sampling
conditions using a representative analyte. Flow, gas temperature and diluent
data are recorded concurrently with the FTIR measurements to provide mass
emission rates for detected compounds.
3.29 Surrogate.
A surrogate is a compound
that is used in a QA spike procedure (section 8.6.2 of this method) to
represent other compounds. The chemical and physical properties of a surrogate
shall be similar to the compounds it is chosen to represent. Under given
sampling conditions, usually a single sampling factor is of primary concern for
measuring the target analytes: for example, the surrogate spike results can be
representative for analytes that are more reactive, more soluble, have a lower
absorptivity, or have a lower vapor pressure than the surrogate itself.
4.0 Interferences.
Interferences are divided
into two classifications: analytical and sampling.
4.1 Analytical Interferences.
An analytical interference
is a spectral feature that complicates (in extreme cases may prevent) the
analysis of an analyte. Analytical interferences are classified as background
or spectral interference.
4.1.1 Background Interference.
This results from a change
in throughput relative to the single beam background. It is corrected by
collecting a new background and proceeding with the test. In severe instances
the cause must be identified and corrected. Potential causes include: (1)
deposits on reflective surfaces or transmitting windows, (2) changes in
detector sensitivity, (3) a change in the infrared source output, or (4)
failure in the instrument electronics. In routine sampling throughput may
degrade over several hours. Periodically a new background must be collected,
but no other corrective action will be required.
4.1.2 Spectral Interference.
This results from the
presence of interfering compound(s) (interferant) in the sample. Interferant
spectral features overlap analyte spectral features. Any compound with an
infrared spectrum, including analytes, can potentially be an interferant. The
Protocol measures absorbance band overlap in each analytical region to
determine if potential interferants shall be classified as known interferants
(FTIR Protocol, section 4.9 and appendix B). Water vapor and CO2 are common spectral interferants. Both of these compounds have strong
infrared spectra and are present in many sample matrices at high concentrations
relative to analytes. The extent of interference depends on the (1) interferant
concentration, (2) analyte concentration, and (3) the degree of band overlap.
Choosing an alternate analytical region can minimize or avoid the spectral
interference. For example, CO2 interferes with the analysis of
the 670 cm-1 benzene band. However, benzene can also be measured
near 3000 cm-1 (with less sensitivity).
4.2 Sampling System Interferences.
These prevent analytes from
reaching the instrument. The analyte spike procedure is designed to measure
sampling system interference, if any.
4.2.1 Temperature. A
temperature that is too low causes condensation of analytes or water vapor. The
materials of the sampling system and the FTIR gas cell usually set the upper
limit of temperature.
4.2.2 Reactive Species.
Anything that reacts with analytes. Some analytes, like formaldehyde,
polymerize at lower temperatures.
4.2.3 Materials. Poor
choice of material for probe, or sampling line may remove some analytes. For
example, HF reacts with glass components.
4.2.4 Moisture. In
addition to being a spectral interferant, condensed moisture removes soluble
compounds.
5.0 Safety.
The hazards of performing
this method are those associated with any stack sampling method and the same
precautions shall be followed. Many HAPs are suspected carcinogens or present
other serious health risks. Exposure to these compounds should be avoided in
all circumstances. For instructions on the safe handling of any particular
compound, refer to its material safety data sheet. When using analyte
standards, always ensure that gases are properly vented and that the gas
handling system is leak free. (Always perform a leak check with the system
under maximum vacuum and, again, with the system at greater than ambient
pressure.) Refer to section 8.2 of this method for leak check procedures. This
method does not address all of the potential safety risks associated with its
use. Anyone performing this method must follow safety and health practices
consistent with applicable legal requirements and with prudent practice for
each application.
6.0 Equipment and Supplies.
Note: Mention of trade names or specific products does not
constitute endorsement by the Environmental Protection Agency.
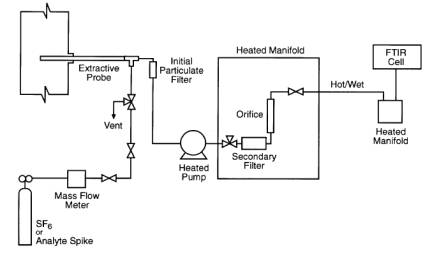
The equipment and supplies
are based on the schematic of a sampling system shown in Figure 1. Either the
batch or continuous sampling procedures may be used with this sampling system.
Alternative sampling configurations may also be used, provided that the data
quality objectives are met as determined in the post-analysis evaluation. Other
equipment or supplies may be necessary, depending on the design of the sampling
system or the specific target analytes.
6.1 Sampling Probe.
Glass, stainless steel, or
other appropriate material of sufficient length and physical integrity to
sustain heating, prevent adsorption of analytes, and to transport analytes to
the infrared gas cell. Special materials or configurations may be required in
some applications. For instance, high stack sample temperatures may require
special steel or cooling the probe. For very high moisture sources it may be
desirable to use a dilution probe.
6.2 Particulate Filters.
A glass wool plug
(optional) inserted at the probe tip (for large particulate removal) and a
filter (required) rated for 99 percent removal efficiency at 1-micron (e.g.,
Balston™) connected at the outlet of the heated probe.
6.3 Sampling Line/Heating System.
Heated (sufficient to
prevent condensation) stainless steel, polytetrafluoroethane, or other material
inert to the analytes.
6.4 Gas Distribution Manifold.
A heated manifold allowing
the operator to control flows of gas standards and samples directly to the FTIR
system or through sample conditioning systems. Usually includes heated flow
meter, heated valve for selecting and sending sample to the analyzer, and a
bypass vent. This is typically constructed of stainless steel tubing and
fittings, and high-temperature valves.
6.5 Stainless Steel Tubing.
Type 316, appropriate
diameter (e.g., 3/8 in.) and length for heated connections. Higher grade
stainless may be desirable in some applications.
6.6 Calibration/Analyte Spike Assembly.
A three way valve assembly
(or equivalent) to introduce analyte or surrogate spikes into the sampling
system at the outlet of the probe upstream of the out-of-stack particulate
filter and the FTIR analytical system.
6.7 Mass Flow Meter (MFM).
These are used for
measuring analyte spike flow. The MFM shall be calibrated in the range of 0 to
5 L/min and be accurate to ± 2 percent (or better) of the flow meter span.
6.8 Gas Regulators.
Appropriate for individual
gas standards.
6.9 Polytetrafluoroethane Tubing.
Diameter (e.g., 3/8 in.)
and length suitable to connect cylinder regulators to gas standard manifold.
6.10 Sample Pump.
A leak-free pump (e.g.,
KNF™), with bypass valve, capable of producing a sample
flow rate of at least 10 L/min through 100 ft of sample line. If the pump is
positioned upstream of the distribution manifold and FTIR system, use a heated
pump that is constructed from materials non-reactive to the analytes. If the
pump is located downstream of the FTIR system, the gas cell sample pressure
will be lower than ambient pressure and it must be recorded at regular
intervals.
6.11 Gas Sample Manifold.
Secondary manifold to
control sample flow at the inlet to the FTIR manifold. This is optional, but
includes a by-pass vent and heated rotameter.
6.12 Rotameter.
A 0 to 20 L/min rotameter.
This meter need not be calibrated.
6.13 FTIR Analytical System.
Spectrometer and detector,
capable of measuring the analytes to the chosen detection limit. The system
shall include a personal computer with compatible software allowing automated
collection of spectra.
6.14 FTIR Cell Pump.
Required for the batch
sampling technique, capable of evacuating the FTIR cell volume within 2
minutes. The pumping speed shall allow the operator to obtain 8 sample spectra
in 1 hour.
6.15 Absolute Pressure Gauge.
Capable of measuring
pressure from 0 to 1000 mmHg to within ± 2.5 mmHg (e.g., Baratron™).
6.16 Temperature Gauge.
Capable of measuring the
cell temperature to within ± 2°C.
6.17 Sample Conditioning.
One option is a condenser
system, which is used for moisture removal. This can be helpful in the
measurement of some analytes. Other sample conditioning procedures may be
devised for the removal of moisture or other interfering species.
6.17.1 The analyte spike
procedure of section 9.2 of this method, the QA spike procedure of section
8.6.2 of this method, and the validation procedure of section 13 of this method
demonstrate whether the sample conditioning affects analyte concentrations.
Alternatively, measurements can be made with two parallel FTIR systems; one
measuring conditioned sample, the other measuring unconditioned sample.
6.17.2 Another option is
sample dilution. The dilution factor measurement must be documented and
accounted for in the reported concentrations. An alternative to dilution is to
lower the sensitivity of the FTIR system by decreasing the cell path length, or
to use a short-path cell in conjunction with a long path cell to measure more
than one concentration range.
7.0 Reagents and Standards.
7.1 Analyte(s) and Tracer Gas.
Obtain a certified gas
cylinder mixture containing all of the analyte(s) at concentrations within ± 2
percent of the emission source levels (expressed in ppm-meter/K). If practical,
the analyte standard cylinder shall also contain the tracer gas at a
concentration which gives a measurable absorbance at a dilution factor of at
least 10:1. Two ppm SF6 is sufficient for a path length of 22 meters at 250
°F.
7.2 Calibration Transfer Standard(s).
Select the calibration
transfer standards (CTS) according to section 4.5 of the FTIR Protocol. Obtain
a National Institute of Standards and Technology (NIST) traceable gravimetric
standard of the CTS (± 2 percent).
7.3 Reference Spectra.
Obtain reference spectra
for each analyte, interferant, surrogate, CTS, and tracer. If EPA reference
spectra are not available, use reference spectra prepared according to procedures
in section 4.6 of the EPA FTIR Protocol.
8.0 Sampling and Analysis Procedure.
Three types of testing can
be performed: (1) screening, (2) emissions test, and (3) validation. Each is
defined in section 3 of this method. Determine the purpose(s) of the FTIR test.
Test requirements include: (a) AUi, DLi, overall fractional uncertainty, OFUi, maximum expected
concentration (CMAXi), and tAN for each, (b)
potential interferants, (c) sampling system factors, e.g., minimum absolute
cell pressure, (Pmin), FTIR cell volume (VSS), estimated
sample absorption pathlength, LS', estimated sample pressure, PS', TS', signal integration time (tSS), minimum instrumental line-width, MIL, fractional error, and (d)
analytical regions, e.g., m = 1 to M, lower wavenumber position, FLm, center wavenumber position, FCm, and upper
wavenumber position, FUm, plus interferants, upper wavenumber position of the
CTS absorption band, FFUm, lower wavenumber position of the CTS absorption
band, FFLm, wavenumber range FNU to FNL. If necessary, sample
and acquire an initial spectrum. From analysis of this preliminary spectrum
determine a suitable operational path length. Set up the sampling train as
shown in Figure 1 or use an appropriate alternative configuration. Sections 8.1
through 8.11 of this method provide guidance on pre-test calculations in the
EPA protocol, sampling and analytical procedures, and post-test protocol
calculations.
8.1 Pretest Preparations and Evaluations.
Using the procedure in
section 4.0 of the FTIR Protocol, determine the optimum sampling system
configuration for measuring the target analytes. Use available information to
make reasonable assumptions about moisture content and other interferences.
8.1.1 Analytes.
Select the required
detection limit (DLi) and the maximum permissible analytical uncertainty
(AUi) for each analyte (labeled from 1 to i). Estimate,
if possible, the maximum expected concentration for each analyte, CMAXi. The expected measurement range is fixed by DLi and CMAXi for each analyte (i).
8.1.2 Potential Interferants.
List the potential
interferants. This usually includes water vapor and CO2, but may also include some analytes and other compounds.
8.1.3. Optical Configuration.
Choose an optical
configuration that can measure all of the analytes within the absorbance range
of .01 to 1.0 (this may require more than one path length). Use Protocol
sections 4.3 to 4.8 for guidance in choosing a configuration and measuring CTS.
8.1.4. Fractional Reproducibility Uncertainty (FRUi).
The FRU is determined for
each analyte by comparing CTS spectra taken before and after the reference
spectra were measured. The EPA para-xylene reference spectra were collected on
10/31/91 and 11/01/91 with corresponding CTS spectra "cts1031a," and "cts1101b."
The CTS spectra are used to estimate the reproducibility (FRU) in the system
that was used to collect the references. The FRU must be < AU. Appendix E of
the protocol is used to calculate the FRU from CTS spectra. Figure 2 plots
results for 0.25 cm-1 CTS spectra in EPA reference library: S3 (cts1101b - cts1031a), and S4 [(cts1101b +
cts1031a)/2]. The RMSD (SRMS) is calculated in the subtracted baseline, S3, in the corresponding CTS region from 850 to 1065 cm-1. The area (BAV) is calculated in the same region of the averaged CTS
spectrum, S4.
8.1.5 Known Interferants.
Use appendix B of the EPA
FTIR Protocol.
8.1.6 Calculate the Minimum Analyte Uncertainty, MAU
(Section 1.3 of this
method discusses MAU and protocol appendix D gives the MAU procedure). The MAU for
each analyte, i, and each analytical region, m, depends on the RMS noise.
8.1.7 Analytical Program.
See FTIR Protocol, section
4.10. Prepare computer program based on the chosen analytical technique. Use as
input reference spectra of all target analytes and expected interferants.
Reference spectra of additional compounds shall also be included in the program
if their presence (even if transient) in the samples is considered possible.
The program output shall be in ppm (or ppb) and shall be corrected for
differences between the reference path length, LR, temperature,
TR, and pressure, PR, and the conditions
used for collecting the sample spectra. If sampling is performed at ambient
pressure, then any pressure correction is usually small relative to corrections
for path length and temperature, and may be neglected.
8.2 Leak-check.
8.2.1 Sampling System. A
typical FTIR extractive sampling train is shown in Figure 1. Leak check from
the probe tip to pump outlet as follows: Connect a 0– to 250-mL/min rate meter
(rotameter or bubble meter) to the outlet of the pump. Close off the inlet to
the probe, and record the leak rate. The leak rate shall be < 200
mL/min.
8.2.2 Analytical System
Leak check. Leak check the FTIR cell under vacuum and under pressure (greater
than ambient). Leak check connecting tubing and inlet manifold under
pressure.
8.2.2.1 For the evacuated
sample technique, close the valve to the FTIR cell, and evacuate the absorption
cell to the minimum absolute pressure Pmin. Close the valve
to the pump, and determine the change in pressure •Pv after 2 minutes.
8.2.2.2 For both the
evacuated sample and purging techniques, pressurize the system to about 100
mmHg above atmospheric pressure.
Isolate the pump and determine the change in pressure •Pp after 2 minutes.
8.2.2.3 Measure the
barometric pressure, Pb in mmHg.
8.2.2.4 Determine the
percent leak volume %VL for the signal integration time tSS and for •Pmax, i.e., the larger of •Pv or •Pp, as follows:

where:
50 = 100% divided by the
leak-check time of 2 minutes.
8.2.2.5 Leak volumes in
excess of 4 percent of the FTIR system volume VSS are
unacceptable.
8.3 Detector Linearity.
Once an optical
configuration is chosen, use one of the procedures of sections 8.3.1 through
8.3.3 to verify that the detector response is linear. If the detector response
is not linear, decrease the aperture, or attenuate the infrared beam. After a
change in the instrument configuration perform a linearity check until it is
demonstrated that the detector response is linear.
8.3.1 Vary the power
incident on the detector by modifying the aperture setting. Measure the
background and CTS at three instrument aperture settings: (1) at the aperture
setting to be used in the testing, (2) at one half this aperture and (3) at
twice the proposed testing aperture. Compare the three CTS spectra. CTS band
areas shall agree to within the uncertainty of the cylinder standard and the
RMSD noise in the system. If test aperture is the maximum aperture, collect CTS
spectrum at maximum aperture, then close the aperture to reduce the IR
throughput by half. Collect a second background and CTS at the smaller aperture
setting and compare the spectra again.
8.3.2 Use neutral density
filters to attenuate the infrared beam. Set up the FTIR system as it will be
used in the test measurements. Collect a CTS spectrum. Use a neutral density
filter to attenuate the infrared beam (either immediately after the source or
the interferometer) to approximately 1/2 its original intensity. Collect a
second CTS spectrum. Use another filter to attenuate the infrared beam to
approximately 1/4 its original intensity. Collect a third background and CTS
spectrum. Compare the CTS spectra. CTS band areas shall agree to within the
uncertainty of the cylinder standard and the RMSD noise in the system.
8.3.3 Observe the single
beam instrument response in a frequency region where the detector response is
known to be zero. Verify that the detector response is "flat" and
equal to zero in these regions.
8.4 Data Storage Requirements.
All field test spectra
shall be stored on a computer disk and a second backup copy must be stored on a
separate disk. The stored information includes sample interferograms, processed
absorbance spectra, background interferograms, CTS sample interferograms and
CTS absorbance spectra. Additionally, documentation of all sample conditions,
instrument settings, and test records must be recorded on hard copy or on
computer medium. Table 1 gives a sample presentation of documentation.
8.5 Background Spectrum.
Evacuate the gas cell to <
5 mmHg, and fill with dry nitrogen gas to ambient pressure (or purge the cell
with 10 volumes of dry nitrogen). Verify that no significant amounts of
absorbing species (for example water vapor and CO2) are present.
Collect a background spectrum, using a signal averaging period equal to or
greater than the averaging period for the sample spectra. Assign a unique file
name to the background spectrum. Store two copies of the background
interferogram and processed single-beam spectrum on separate computer disks
(one copy is the back-up).
8.5.1 Interference
Spectra. If possible, collect spectra of known and suspected major
interferences using the same optical system that will be used in the field
measurements. This can be done on-site or earlier. A number of gases, e.g. CO2, SO2, CO, NH3, are readily
available from cylinder gas suppliers.
8.5.2 Water vapor spectra
can be prepared by the following procedure. Fill a sample tube with distilled
water. Evacuate above the sample and remove dissolved gasses by alternately
freezing and thawing the water while evacuating. Allow water vapor into the
FTIR cell, then dilute to atmospheric pressure with nitrogen or dry air. If
quantitative water spectra are required, follow the reference spectrum
procedure for neat samples (protocol, section 4.6). Often, interference spectra
need not be quantitative, but for best results the absorbance must be
comparable to the interference absorbance in the sample spectra.
8.6 Pre-Test Calibrations
8.6.1 Calibration Transfer Standard.
Evacuate the gas cell to <
5 mmHg absolute pressure, and fill the FTIR cell to atmospheric pressure
with the CTS gas. Alternatively, purge the cell with 10 cell volumes of CTS
gas. (If purge is used, verify that the CTS concentration in the cell is stable
by collecting two spectra 2 minutes apart as the CTS gas continues to flow. If
the absorbance in the second spectrum is no greater than in the first, within
the uncertainty of the gas standard, then this can be used as the CTS
spectrum.) Record the spectrum.
8.6.2 QA Spike.
This procedure assumes
that the method has been validated for at least some of the target analytes at
the source. For emissions testing perform a QA spike. Use a certified standard,
if possible, of an analyte, which has been validated at the source. One analyte
standard can serve as a QA surrogate for other analytes which are less reactive
or less soluble than the standard. Perform the spike procedure of section 9.2
of this method. Record spectra of at least three independent (section 3.22 of
this method) spiked samples. Calculate the spiked component of the analyte
concentration. If the average spiked concentration is within 0.7 to 1.3 times
the expected concentration, then proceed with the testing. If applicable, apply
the correction factor from the Method 301 of this appendix validation test (not
the result from the QA spike).
8.7 Sampling.
If analyte concentrations
vary rapidly with time, continuous sampling is preferable using the smallest
cell volume, fastest sampling rate and fastest spectra collection rate
possible. Continuous sampling requires the least operator intervention even
without an automated sampling system. For continuous monitoring at one location
over long periods, Continuous sampling is preferred. Batch sampling and
continuous static sampling are used for screening and performing test runs of
finite duration. Either technique is preferred for sampling several locations
in a matter of days. Batch sampling gives reasonably good time resolution and
ensures that each spectrum measures a discreet (and unique) sample volume.
Continuous static (and continuous) sampling provides a very stable background
over long periods. Like batch sampling, continuous static sampling also ensures
that each spectrum measures a unique sample volume. It is essential that the
leak check procedure under vacuum (section 8.2 of this method) is passed if the
batch sampling procedure is used. It is essential that the leak check procedure
under positive pressure is passed if the continuous static or continuous
sampling procedures are used. The sampling techniques are described in sections
8.7.1 through 8.7.2 of this method.
8.7.1 Batch Sampling.
Evacuate the absorbance cell to < 5 mmHg absolute pressure. Fill the
cell with exhaust gas to ambient pressure, isolate the cell, and record the
spectrum. Before taking the next sample, evacuate the cell until no spectral
evidence of sample absorption remains. Repeat this procedure to collect eight
spectra of separate samples in 1 hour.
8.7.2 Continuous Static
Sampling. Purge the FTIR cell with 10 cell volumes of sample gas. Isolate the
cell, collect the spectrum of the static sample and record the pressure. Before
measuring the next sample, purge the cell with 10 more cell volumes of sample
gas.
8.8 Sampling QA and Reporting.
8.8.1 Sample integration
times shall be sufficient to achieve the required signal-to-noise ratio. Obtain
an absorbance spectrum by filling the cell with N2. Measure the
RMSD in each analytical region in this absorbance spectrum. Verify that the
number of scans used is sufficient to achieve the target MAU.
8.8.2 Assign a unique file
name to each spectrum.
8.8.3 Store two copies of
sample interferograms and processed spectra on separate computer disks.
8.8.4 For each sample
spectrum, document the sampling conditions, the sampling time (while the cell
was being filled), the time the spectrum was recorded, the instrumental
conditions (path length, temperature, pressure, resolution, signal integration
time), and the spectral file name. Keep a hard copy of these data sheets.
8.9 Signal Transmittance.
While sampling, monitor
the signal transmittance. If signal transmittance (relative to the background)
changes by 5 percent or more (absorbance =
-.02 to .02) in any analytical spectral region, obtain a new background
spectrum.
8.10 Post-test CTS.
After the sampling run,
record another CTS spectrum.
8.11 Post-test QA.
8.11.1 Inspect the sample
spectra immediately after the run to verify that the gas matrix composition was
close to the expected (assumed) gas matrix.
8.11.2 Verify that the
sampling and instrumental parameters were appropriate for the conditions
encountered. For example, if the moisture is much greater than anticipated, it
may be necessary to use a shorter path length or dilute the sample.
8.11.3 Compare the pre-
and post-test CTS spectra. The peak absorbance in pre- and post-test CTS must
be ± 5 percent of the mean value. See appendix E of the FTIR Protocol.
9.0 Quality Control.
Use analyte spiking
(sections 8.6.2, 9.2 and 13.0 of this method) to verify that the sampling
system can transport the analytes from the probe to the FTIR system.
9.1 Spike Materials.
Use a certified standard
(accurate to ± 2 percent) of the target analyte, if one can be obtained. If a
certified standard cannot be obtained, follow the procedures in section 4.6.2.2
of the FTIR Protocol.
9.2 Spiking Procedure.
QA spiking (section 8.6.2
of this method) is a calibration procedure used before testing. QA spiking involves
following the spike procedure of sections 9.2.1 through 9.2.3 of this method to
obtain at least three spiked samples. The analyte concentrations in the spiked
samples shall be compared to the expected spike concentration to verify that
the sampling/analytical system is working properly. Usually, when QA spiking is
used, the method has already been validated at a similar source for the analyte
in question. The QA spike demonstrates that the validated sampling/analytical
conditions are being duplicated. If the QA spike fails then the
sampling/analytical system shall be repaired before testing proceeds. The
method validation procedure (section 13.0 of this method) involves a more
extensive use of the analyte spike procedure of sections 9.2.1 through 9.2.3 of
this method. Spectra of at least 12 independent spiked and 12 independent
unspiked samples are recorded. The concentration results are analyzed
statistically to determine if there is a systematic bias in the method for
measuring a particular analyte. If there is a systematic bias, within the
limits allowed by Method 301 of this appendix, then a correction factor shall
be applied to the analytical results. If the systematic bias is greater than
the allowed limits, this method is not valid and cannot be used.
9.2.1 Introduce the
spike/tracer gas at a constant flow rate of < 10 percent of the total
sample flow, when possible. (Note: Use the rotameter at the end of the sampling
train to estimate the required spike/tracer gas flow rate.) Use a flow device,
e.g., mass flow meter (± 2 percent), to monitor the spike flow rate. Record the
spike flow rate every 10 minutes.
9.2.2 Determine the
response time (RT) of the system by continuously collecting spectra of the
spiked effluent until the spectrum of the spiked component is constant for 5
minutes. The RT is the interval from the first measurement until the spike
becomes constant. Wait for twice the duration of the RT, then collect spectra
of two independent spiked gas samples. Duplicate analyses of the spiked concentration
shall be within 5 percent of the mean of the two measurements.
9.2.3 Calculate the
dilution ratio using the tracer gas as follows:

where:
![]()


10.0 Calibration and Standardization.
10.1 Signal-to-Noise Ratio (S/N).
The RMSD in the noise must
be less than one tenth of the minimum analyte peak absorbance in each
analytical region. For example if the minimum peak absorbance is 0.01 at the
required DL, then RMSD measured over the entire analytical region must be <
0.001.
10.2 Absorbance Path length.
Verify the absorbance path
length by comparing reference CTS spectra to test CTS spectra. See appendix E
of the FTIR Protocol.
10.3 Instrument Resolution.
Measure the line width of
appropriate test CTS band(s) to verify instrument resolution. Alternatively,
compare CTS spectra to a reference CTS spectrum, if available, measured at the
nominal resolution.
10.4 Apodization Function.
In transforming the sample
interferograms to absorbance spectra use the same apodization function that was
used in transforming the reference spectra.
10.5 FTIR Cell Volume.
Evacuate the cell to <
5 mmHg. Measure the initial absolute temperature (Ti) and absolute pressure (Pi). Connect a wet
test meter (or a calibrated dry gas meter), and slowly draw room air into the
cell. Measure the meter volume (Vm), meter absolute
temperature (Tm), and meter absolute pressure (Pm); and the cell final absolute temperature (Tf) and absolute pressure (Pf). Calculate the
FTIR cell volume VSS, including that of the connecting tubing, as
follows:

11.0 Data Analysis and Calculations.
Analyte concentrations
shall be measured using reference spectra from the EPA FTIR spectral library.
When EPA library spectra are not available, the procedures in section 4.6 of
the Protocol shall be followed to prepare reference spectra of all the target
analytes.
11.1 Spectral De-resolution.
Reference spectra can be
converted to lower resolution standard spectra (section 3.3 of this method) by
truncating the original reference sample and background interferograms.
Appendix K of the FTIR Protocol gives specific deresolution procedures.
Deresolved spectra shall be transformed using the same apodization function and
level of zero filling as the sample spectra. Additionally, pre-test FTIR protocol
calculations (e.g., FRU, MAU, FCU) shall be performed using the de-resolved
standard spectra.
11.2 Data Analysis.
Various analytical
programs are available for relating sample absorbance to a concentration
standard. Calculated concentrations shall be verified by analyzing residual
baselines after mathematically subtracting scaled reference spectra from the
sample spectra. A full description of the data analysis and calculations is
contained in the FTIR Protocol (sections 4.0, 5.0, 6.0 and appendices). Correct
the calculated concentrations in the sample spectra for differences in
absorption path length and temperature between the reference and sample spectra
using equation 6,

where:


12.0 Method Performance.
12.1 Spectral Quality.
Refer to the FTIR Protocol
appendices for analytical requirements, evaluation of data quality, and
analysis of uncertainty.
12.2 Sampling QA/QC.
The analyte spike
procedure of section 9 of this method, the QA spike of section 8.6.2 of this
method, and the validation procedure of section 13 of this method are used to
evaluate the performance of the sampling system and to quantify sampling system
effects, if any, on the measured concentrations. This method is self-validating
provided that the results meet the performance requirement of the QA spike in
sections 9.0 and 8.6.2 of this method and results from a previous method
validation study support the use of this method in the application. Several
factors can contribute to uncertainty in the measurement of spiked samples.
Factors which can be controlled to provide better accuracy in the spiking
procedure are listed in sections 12.2.1 through 12.2.4 of this method.
12.2.1 Flow meter. An
accurate mass flow meter is accurate to ± 1 percent of its span. If a flow of 1
L/min is monitored with such a MFM, which is calibrated in the range of 0-5
L/min, the flow measurement has an uncertainty of 5 percent. This may be
improved by re-calibrating the meter at the specific flow rate to be used.
12.2.2 Calibration gas.
Usually the calibration standard is certified to within ± 2 percent. With
reactive analytes, such as HCl, the certified accuracy in a commercially
available standard may be no better than ± 5 percent.
12.2.3 Temperature.
Temperature measurements of the cell shall be quite accurate. If practical, it
is preferable to measure sample temperature directly, by inserting a
thermocouple into the cell chamber instead of monitoring the cell outer wall
temperature.
12.2.4 Pressure. Accuracy
depends on the accuracy of the barometer, but fluctuations in pressure
throughout a day may be as much as 2.5 percent due to weather variations.
13.0 Method Validation Procedure.
This validation procedure,
which is based on EPA Method 301 (40 CFR part 63, appendix A), may be used to
validate this method for the analytes in a gas matrix. Validation at one source
may also apply to another type of source, if it can be shown that the exhaust
gas characteristics are similar at both sources.
13.1 Analyte Spike procedure
Section 5.3 of Method 301
(40 CFR part 63, appendix A), the Analyte Spike procedure, is used with these
modifications. The statistical analysis of the results follows section 6.3 of
EPA Method 301. Section 3 of this method defines terms that are not defined in
Method 301.
13.1.1 The analyte spike
is performed dynamically. This means the spike flow is continuous and constant
as spiked samples are measured.
13.1.2 The spike gas is
introduced at the back of the sample probe.
13.1.3 Spiked effluent is
carried through all sampling components downstream of the probe.
13.1.4 A single FTIR
system (or more) may be used to collect and analyze spectra (not quadruplicate
integrated sampling trains).
13.1.5 All of the
validation measurements are performed sequentially in a single "run"
(section 3.26 of this method).
13.1.6 The measurements
analyzed statistically are each independent (section 3.22 of this method).
13.1.7 A validation data
set can consist of more than 12 spiked and 12 unspiked measurements.
13.2 Batch Sampling.
The procedure in sections
13.2.1 through 13.2.2 may be used for stable processes. If process emissions
are highly variable, the procedure in section
13.2.3 shall be used.
13.2.1 With a single FTIR
instrument and sampling system, begin by collecting spectra of two unspiked
samples. Introduce the spike flow into the sampling system and allow 10 cell
volumes to purge the sampling system and FTIR cell. Collect spectra of two
spiked samples. Turn off the spike and allow 10 cell volumes of unspiked sample
to purge the FTIR cell. Repeat this procedure until the 24 (or more) samples
are collected.
13.2.2 In batch sampling,
collect spectra of 24 distinct samples. (Each distinct sample consists of
filling the cell to ambient pressure after the cell has been evacuated.)
13.2.3 Alternatively, a
separate probe assembly, line, and sample pump can be used for spiked sample.
Verify and document that sampling conditions are the same in both the spiked
and the unspiked sampling systems. This can be done by wrapping both sample
lines in the same heated bundle. Keep the same flow rate in both sample lines.
Measure samples in sequence in pairs. After two spiked samples are measured,
evacuate the FTIR cell, and turn the manifold valve so that spiked sample flows
to the FTIR cell. Allow the connecting line from the manifold to the FTIR cell
to purge thoroughly (the time depends on the line length and flow rate).
Collect a pair of spiked samples. Repeat the procedure until at least 24
measurements are completed.
13.3 Simultaneous Measurements With Two FTIR Systems.
If unspiked effluent
concentrations of the target analyte(s) vary significantly with time, it may be
desirable to perform synchronized measurements of spiked and unspiked sample.
Use two FTIR systems, each with its own cell and sampling system to perform
simultaneous spiked and unspiked measurements. The optical configurations shall
be similar, if possible. The sampling configurations shall be the same. One
sampling system and FTIR analyzer shall be used to measure spiked effluent. The
other sampling system and FTIR analyzer shall be used to measure unspiked flue
gas. Both systems shall use the same sampling procedure (i.e., batch or
continuous).
13.3.1 If batch sampling
is used, synchronize the cell evacuation, cell filling, and collection of
spectra. Fill both cells at the same rate (in cell volumes per unit time).
13.3.2 If continuous
sampling is used, adjust the sample flow through each gas cell so that the same
number of cell volumes pass through each cell in a given time (i.e. TC1 = TC2).
13.4 Statistical Treatment.
The statistical procedure
of EPA Method 301 of this appendix, section 6.3 is used to evaluate the bias
and precision. For FTIR testing a validation "run" is defined as
spectra of 24 independent samples, 12 of which are spiked with the analyte(s)
and 12 of which are not spiked.
13.4.1 Bias. Determine the
bias (defined by EPA Method 301 of this appendix, section 6.3.2) using equation
7:
![]()
where:

13.4.2 Correction Factor.
Use section 6.3.2.2 of Method 301 of this appendix to evaluate the statistical
significance of the bias. If it is determined that the bias is significant,
then use section 6.3.3 of Method 301 to calculate a correction factor (CF).
Analytical results of the test method are multiplied by the correction factor,
if 0.7 < CF < 1.3. If is determined that the bias is
significant and CF > ± 30 percent, then the test method is considered to
"not valid."
13.4.3 If measurements do
not pass validation, evaluate the sampling system, instrument configuration,
and analytical system to determine if improper set-up or a malfunction was the
cause. If so, repair the system and repeat the validation.
14.0 Pollution Prevention.
The extracted sample gas
is vented outside the enclosure containing the FTIR system and gas manifold
after the analysis. In typical method applications the vented sample volume is
a small fraction of the source volumetric flow and its composition is identical
to that emitted from the source. When analyte spiking is used, spiked
pollutants are vented with the extracted sample gas. Approximately 1.6 x 10-4 to 3.2 x 10-4 lbs of a single HAP may be vented to the atmosphere
in a typical validation run of 3 hours. (This assumes a molar mass of 50 to 100
g, spike rate of 1.0 L/min, and a standard concentration of 100 ppm). Minimize
emissions by keeping the spike flow off when not in use.
15.0 Waste Management.
Small volumes of
laboratory gas standards can be vented through a laboratory hood. Neat samples
must be packed and disposed according to applicable regulations. Surplus
materials may be returned to supplier for disposal.
16.0 References.
1. "Field Validation
Test Using Fourier Transform Infrared (FTIR) Spectrometry To Measure
Formaldehyde, Phenol and Methanol at a Wool Fiberglass Production
Facility." Draft. U.S. Environmental Protection Agency Report, EPA
Contract No. 68D20163, Work Assignment I-32, September 1994.
2. "FTIR Method
Validation at a Coal-Fired Boiler". Prepared for U.S. Environmental
Protection Agency, Research Triangle Park, NC. Publication No.:
EPA-454/R95-004, NTIS No.: PB95-193199. July, 1993.
3. "Method 301 -
Field Validation of Pollutant Measurement Methods from Various Waste
Media," 40 CFR part 63, appendix A.
4. "Molecular
Vibrations; The Theory of Infrared and Raman Vibrational Spectra," E.
Bright Wilson, J. C. Decius, and P. C. Cross, Dover Publications, Inc., 1980.
For a less intensive treatment of molecular rotational-vibrational spectra see,
for example, "Physical Chemistry," G. M. Barrow, chapters 12, 13, and
14, McGraw Hill, Inc., 1979.
5. "Fourier Transform
Infrared Spectrometry," Peter R. Griffiths and James de Haseth, Chemical
Analysis, 83, 16-25,(1986), P. J. Elving, J. D. Winefordner and
I. M. Kolthoff (ed.), John Wiley and Sons.
6. "Computer-Assisted
Quantitative Infrared Spectroscopy," Gregory L. McClure (ed.), ASTM
Special Publication 934 (ASTM),
1987.
7. "Multivariate
Least-Squares Methods Applied to the Quantitative Spectral Analysis of
Multicomponent Mixtures," Applied Spectroscopy, 39(10),
73-84, 1985.
Table 1. EXAMPLE
PRESENTATION OF SAMPLING DOCUMENTATION.

Figure 1. Extractive
FTIR sampling system.
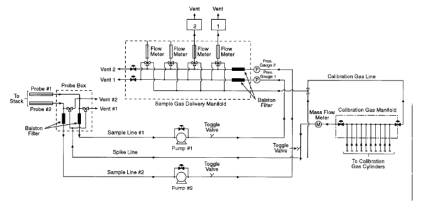
Figure 2. Fractional Reproducibility. Top: average of cts1031a and cts1101b. Bottom: Reference spectrum of p-xylene.
ADDENDUM TO TEST METHOD
320 PROTOCOL FOR THE USE OF EXTRACTIVE FOURIER TRANSFORM INFRARED (FTIR)
SPECTROMETRY FOR THE ANALYSES OF GASEOUS EMISSIONS FROM STATIONARY SOURCES
1.0 INTRODUCTION
The purpose of this
addendum is to set general guidelines for the use of modern FTIR spectroscopic
methods for the analysis of gas samples extracted from the effluent of
stationary emission sources. This addendum outlines techniques for developing
and evaluating such methods and sets basic requirements for reporting and
quality assurance procedures.
1.1 NOMENCLATURE
1.1.1 Appendix A to this
addendum lists definitions of the symbols and terms used in this Protocol, many
of which have been taken directly from American Society for Testing and
Materials (ASTM) publication E 131-90a, entitled "Terminology Relating to
Molecular Spectroscopy."
1.1.2 Except in the case
of background spectra or where otherwise noted, the term "spectrum"
refers to a double-beam spectrum in units of absorbance vs. wavenumber (cm-1).
1.1.3 The term
"Study" in this addendum refers to a publication that has been
subjected to EPA- or peer-review.
2.0 APPLICABILITY AND ANALYTICAL PRINCIPLE
2.1 Applicability.
This Protocol applies to
the determination of compound-specific concentrations in single and
multiple-component gas phase samples using double-beam absorption spectroscopy
in the mid-infrared band. It does not specifically address other FTIR
applications, such as single-beam spectroscopy, analysis of open-path
(non-enclosed) samples, and continuous measurement techniques. If multiple
spectrometers, absorption cells, or instrumental linewidths are used in such
analyses, each distinct operational configuration of the system must be
evaluated separately according to this Protocol.
2.2 Analytical Principle.
2.2.1 In the mid-infrared
band, most molecules exhibit characteristic gas phase absorption spectra that
may be recorded by FTIR systems. Such systems consist of a source of
mid-infrared radiation, an interferometer, an enclosed sample cell of known
absorption pathlength, an infrared detector, optical elements for the transfer
of infrared radiation between components, and gas flow control and measurement
components. Adjunct and integral computer systems are used for controlling the
instrument, processing the signal, and for performing both Fourier transforms
and quantitative analyses of spectral data.
2.2.2 The absorption
spectra of pure gases and of mixtures of gases are described by a linear
absorbance theory referred to as Beer's Law. Using this law, modern FTIR
systems use computerized analytical programs to quantify compounds by comparing
the absorption spectra of known (reference) gas samples to the absorption
spectrum of the sample gas. Some standard mathematical techniques used for
comparisons are classical least squares, inverse least squares,
cross-correlation, factor analysis, and partial least squares. Reference A
describes several of these techniques, as well as additional techniques, such
as differentiation methods, linear baseline corrections, and
non-linear absorbance
corrections.
3.0 GENERAL PRINCIPLES OF PROTOCOL REQUIREMENTS
The characteristics that
distinguish FTIR systems from gas analyzers used in instrumental gas analysis
methods (e.g., Methods 6C and 7E of appendix A to part 60 of this chapter) are:
(1) Computers are necessary to obtain and analyze data; (2) chemical
concentrations can be quantified using previously recorded infrared reference
spectra; and (3) analytical assumptions and results, including possible effects
of interfering compounds, can be evaluated after the quantitative analysis. The
following general principles and requirements of this Protocol are based on
these characteristics.
3.1 Verifiability and Reproducibility of Results.
Store all data and
document data analysis techniques sufficient to allow an independent agent to
reproduce the analytical results from the raw interferometric data.
3.2 Transfer of Reference Spectra.
To determine whether
reference spectra recorded under one set of conditions (e.g., optical bench, instrumental
line-width, absorption pathlength, detector performance, pressure, and
temperature) can be used to analyze sample spectra taken under a different set
of conditions, quantitatively compare "calibration transfer
standards" (CTS) and reference spectra as described in this Protocol.
(Note: The CTS may, but need not, include analytes of interest). To effect
this, record the absorption spectra of the CTS (a) immediately before and
immediately after recording reference spectra and (b) immediately after
recording sample spectra.
3.3 Evaluation of FTIR Analyses.
The applicability,
accuracy, and precision of FTIR measurements are influenced by a number of
interrelated factors, which may be divided into two classes:
3.3.1 Sample-Independent Factors.
Examples are system
configuration and performance (e.g., detector sensitivity and infrared source
output), quality and applicability of reference absorption spectra, and type of
mathematical analyses of the spectra. These factors define the fundamental limitations
of FTIR measurements for a given system configuration. These limitations may be
estimated from evaluations of the system before samples are available. For
example, the detection limit for the absorbing compound under a given set of
conditions may be estimated from the system noise level and the strength of a
particular absorption band. Similarly, the accuracy of measurements may be
estimated from the analysis of the reference spectra.
3.3.2 Sample-Dependent Factors.
Examples are spectral
interferants (e.g., water vapor and CO2) or the overlap of
spectral features of different compounds and contamination deposits on
reflective surfaces or transmitting windows. To maximize the effectiveness of
the mathematical techniques used in spectral analysis, identification of
interferants (a standard initial step) and analysis of samples (includes effect
of other analytical errors) are necessary. Thus, the Protocol requires
post-analysis calculation of measurement concentration uncertainties for the
detection of these potential sources of measurement error.
4.0 PRE-TEST PREPARATIONS AND EVALUATIONS
Before testing,
demonstrate the suitability of FTIR spectrometry for the desired application
according to the procedures of this section.
4.1 Identify Test Requirements.
Identify and record the
test requirements described in sections 4.1.1 through 4.1.4 of this addendum.
These values set the desired or required goals of the proposed analysis; the
description of methods for determining whether these goals are actually met
during the analysis comprises the majority of this Protocol.
4.1.1 Analytes (specific
chemical species) of interest. Label the analytes from i = 1 to I.
4.1.2 Analytical
uncertainty limit (AUi). The AUi is the maximum
permissible fractional uncertainty of analysis for the ith analyte concentration, expressed as a fraction of the analyte
concentration in the sample.
4.1.3 Required detection
limit for each analyte (DLi, ppm). The detection limit is the lowest
concentration of an analyte for which its overall fractional uncertainty (OFUi) is required to be less than its analytical uncertainty limit (AUi).
4.1.4 Maximum expected
concentration of each analyte (CMAXi, ppm).
4.2 Identify Potential Interferants.
Considering the chemistry
of the process or results of previous studies, identify potential interferants,
i.e., the major effluent constituents and any relatively minor effluent
constituents that possess either strong absorption characteristics or strong
structural similarities to any analyte of interest. Label them 1 through Nj, where the subscript "j" pertains to potential interferants.
Estimate the concentrations of these compounds in the effluent (CPOTj, ppm).
4.3 Select and Evaluate the Sampling System.
Considering the source,
e.g., temperature and pressure profiles, moisture content, analyte
characteristics, and particulate concentration), select the equipment for
extracting gas samples. Recommended are a particulate filter, heating system to
maintain sample temperature above the dew point for all sample constituents at
all points within the sampling system (including the filter), and sample
conditioning system (e.g., coolers, water-permeable membranes that remove water
or other compounds from the sample, and dilution devices) to remove spectral
interferants or to protect the sampling and analytical components. Determine
the minimum absolute sample system pressure (Pmin, mmHg) and
the infrared absorption cell volume (VSS, liter). Select
the techniques and/or equipment for the measurement of sample pressures and
temperatures.
4.4 Select Spectroscopic System.
Select a spectroscopic
configuration for the application. Approximate the absorption pathlength (LS', meter), sample pressure (PS', kPa), absolute
sample temperature TS', and signal integration period (tSS, seconds) for the analysis. Specify the nominal minimum instrumental
linewidth (MIL) of the system. Verify that the fractional error at the
approximate values PS' and TS' is less than one
half the smallest value AUi
(see section 4.1.2 of this addendum).
4.5 Select Calibration Transfer Standards (CTS's).
Select CTS's that meet the
criteria listed in sections 4.5.1, 4.5.2, and 4.5.3 of this addendum.
Note: It may be necessary
to choose preliminary analytical regions (see section 4.7 of this addendum),
identify the minimum analyte line-widths, or estimate the system noise level
(see section 4.12 of this addendum) before selecting the CTS. More than one
compound may be needed to meet the criteria; if so, obtain separate cylinders
for each compound.
4.5.1 The central
wavenumber position of each analytical region shall lie within 25 percent of
the wavenumber position of at least one CTS absorption band.
4.5.2 The absorption bands
in section 4.5.1 of this addendum shall exhibit peak absorbances greater than
ten times the value RMSEST
(see section 4.12 of this addendum) but
less than 1.5 absorbance units.
4.5.3 At least one
absorption CTS band within the operating range of the FTIR instrument shall
have an instrument-independent line-width no greater than the narrowest analyte
absorption band. Perform and document measurements or cite Studies to determine
analyte and CTS compound line-widths.
4.5.4 For each analytical
region, specify the upper and lower wavenumber positions (FFUm and FFLm, respectively) that bracket the CTS absorption band
or bands for the associated analytical region. Specify the wavenumber range,
FNU to FNL, containing the absorption band that meets the criterion of section
4.5.3 of this addendum.
4.5.5 Associate, whenever
possible, a single set of CTS gas cylinders with a set of reference spectra.
Replacement CTS gas cylinders shall contain the same compounds at
concentrations within 5 percent of that of the original CTS cylinders; the
entire absorption spectra (not individual spectral segments) of the replacement
gas shall be scaled by a factor between 0.95 and 1.05 to match the original CTS
spectra.
4.6 Prepare Reference Spectra.
Note: Reference spectra
are available in a permanent soft copy from the EPA spectral library on the
EMTIC (Emission Measurement Technical Information Center) computer bulletin
board; they may be used if applicable.
4.6.1 Select the reference
absorption pathlength (LR) of the cell.
4.6.2 Obtain or prepare a
set of chemical standards for each analyte, potential and known spectral
interferants, and CTS. Select the concentrations of the chemical standards to
correspond to the top of the desired range.
4.6.2.1
Commercially-Prepared Chemical Standards. Chemical standards for many compounds
may be obtained from independent sources, such as a specialty gas manufacturer,
chemical company, or commercial laboratory. These standards (accurate to within
±2 percent) shall be prepared according to EPA Traceability Protocol (see
Reference D) or shall be traceable to NIST standards. Obtain from the supplier
an estimate of the stability of the analyte concentration. Obtain and follow
all of the supplier's recommendations for recertifying the analyte
concentration.
4.6.2.2 Self-Prepared
Chemical Standards. Chemical standards may be prepared by diluting certified
commercially prepared chemical gases or pure analytes with ultra-pure carrier
(UPC) grade nitrogen according to the barometric and volumetric techniques
generally described in Reference A, section A4.6.
4.6.3 Record a set of the
absorption spectra of the CTS {R1}, then a set of the reference spectra at two
or more concentrations in duplicate over the desired range (the top of the
range must be less than 10 times that of the bottom), followed by a second set of
CTS spectra {R2}. (If self-prepared standards are used, see section 4.6.5 of
this addendum before disposing of any of the standards.) The maximum accepted
standard concentration-pathlength product (ASCPP) for each compound shall be
higher than the maximum estimated concentration-pathlength products for both
analytes and known interferants in the effluent gas. For each analyte, the
minimum ASCPP shall be no greater than ten times the concentration-pathlength
product of that analyte at its required detection limit.
4.6.4 Permanently store
the background and interferograms in digitized form. Document details of the
mathematical process for generating the spectra from these interferograms.
Record the sample pressure (PR), sample temperature (TR), reference absorption pathlength (LR), and interferogram
signal integration period (tSR). Signal integration periods
for the background interferograms shall be >tSR. Values of PR, LR, and tSR shall not deviate by more than ±1 percent from the time of recording
{R1} to that of recording {R2}.
4.6.5 If self-prepared
chemical standards are employed and spectra of only two concentrations are
recorded for one or more compounds, verify the accuracy of the dilution
technique by analyzing the prepared standards for those compounds with a
secondary (non-FTIR) technique in accordance with sections 4.6.5.1 through
4.6.5.4 of this addendum.
4.6.5.1 Record the
response of the secondary technique to each of the four standards prepared.
4.6.5.2 Perform a linear
regression of the response values (dependant variable) versus the accepted
standard concentration (ASC) values (independent variable), with the regression
constrained to pass through the zero-response, zero ASC point.
4.6.5.3 Calculate the
average fractional difference between the actual response values and the
regression predicted values (those calculated from the regression line using
the four ASC values as the independent variable).
4.6.5.4 If the average
fractional difference value calculated in section 4.6.5.3 of this addendum is
larger for any compound than the corresponding AUi, the dilution
technique is not sufficiently accurate and the reference spectra prepared are
not valid for the analysis.
4.7 Select Analytical Regions.
Using the general
considerations in section 7 of Reference A and the spectral characteristics of
the analytes and interferants, select the analytical regions for the
application. Label them m = 1 to M. Specify the lower, center and upper
wavenumber positions of each analytical region (FLm, FCm, and FUm, respectively).
Specify the analytes and interferants which exhibit absorption in each region.
4.8 Determine Fractional Reproducibility Uncertainties.
Using appendix E of this
addendum, calculate the fractional reproducibility uncertainty for each analyte
(FRUi) from a comparison of {R1} and {R2}. If FRUi > AUi for any analyte, the reference spectra generated in
accordance with section 4.6 of this addendum are not valid for the application.
4.9 Identify Known Interferants.
Using appendix B of this addendum,
determine which potential interferants affect the analyte concentration
determinations. Relabel these potential interferant as "known"
interferants, and designate these compounds from k = 1 to K. Appendix B to this
addendum also provides criteria for determining whether the selected analytical
regions are suitable.
4.10 Prepare Computerized Analytical Programs.
4.10.1 Choose or devise
mathematical techniques (e.g, classical least squares, inverse least squares,
cross-correlation, and factor analysis) based on equation 4 of Reference A that
are appropriate for analyzing spectral data by comparison with reference
spectra.
4.10.2 Following the
general recommendations of Reference A, prepare a computer program or set of
programs that analyzes all of the analytes and known interferants, based on the
selected analytical regions (section 4.7 of this addendum) and the prepared
reference spectra (section 4.6 of this addendum). Specify the baseline
correction technique (e.g., determining the slope and intercept of a linear
baseline contribution in each analytical region) for each analytical region,
including all relevant wavenumber positions.
4.10.3 Use programs that
provide as output [at the reference absorption pathlength (LR), reference gas temperature (TR), and reference gas
pressure (PR)] the analyte concentrations, the known interferant
concentrations, and the baseline slope and intercept values. If the sample
absorption pathlength (LS), sample gas temperature (TS), or sample gas pressure (PS) during the actual
sample analyses differ from LR, TR, and PR, use a program or set of programs that applies
multiplicative corrections to the derived concentrations to account for these
variations, and that provides as output both the corrected and uncorrected values.
Include in the report of the analysis (see section 7.0 of this addendum) the
details of any transformations applied to the original reference spectra (e.g.,
differentiation), in such a fashion that all analytical results may be verified
by an independent agent from the reference spectra and data spectra alone.
4.11 Determine the Fractional Calibration Uncertainty.
Calculate the fractional
calibration uncertainty for each analyte (FCUi) according to appendix F of this addendum, and compare these values to the fractional
uncertainty limits (AUi; see section 4.1.2 of this addendum). If FCUi > AUi, either the reference spectra or analytical programs
for that analyte are unsuitable.
4.12 Verify System Configuration Suitability.
Using appendix C of this addendum,
measure or obtain estimates of the noise level (RMSEST, absorbance) of the FTIR system. Alternatively, construct the complete
spectrometer system and determine the values RMSSm using
appendix G of this addendum. Estimate the minimum measurement uncertainty for
each analyte (MAUi, ppm) and known interferant (MIUk, ppm) using appendix D of this addendum. Verify that (a) MAUi < (AUi)(DLi), FRUi < AUi, and FCUi < AUi for each analyte and that (b) the CTS chosen meets the requirements
listed in sections 4.5.1 through 4.5.5 of this addendum.
5.0 SAMPLING AND ANALYSIS PROCEDURE
5.1 Analysis System Assembly and Leak-Test.
Assemble the analysis
system. Allow sufficient time for all system components to reach the desired
temperature. Then, determine the leak-rate (LR) and leak
volume (VL), where VL = LR tSS. Leak volumes shall be #4 percent of VSS.
5.2 Verify Instrumental Performance.
Measure the noise level of
the system in each analytical region using the procedure of appendix G of this
addendum. If any noise level is higher than that estimated for the system in
section 4.12 of this addendum, repeat the calculations of appendix D of this
addendum and verify that the requirements of section 4.12 of this addendum are
met; if they are not, adjust or repair the instrument and repeat this section.
5.3 Determine the Sample Absorption Pathlength.
Record a background
spectrum. Then, fill the absorption cell with CTS at the pressure PR and record a set of CTS spectra {R3}. Store the background and unscaled
CTS single beam interferograms and spectra. Using appendix H of this addendum,
calculate the sample absorption pathlength (LS) for each
analytical region. The values LS shall not differ from the
approximated sample pathlength LS' (see section 4.4 of this addendum)
by more than 5 percent.
5.4 Record Sample Spectrum.
Connect the sample line to
the source. Either evacuate the absorption cell to an absolute pressure below 5
mmHg before extracting a sample from the effluent stream into the absorption
cell, or pump at least ten cell volumes of sample through the cell before
obtaining a sample. Record the sample pressure PS. Generate the
absorbance spectrum of the sample. Store the background and sample single beam
interferograms, and document the process by which the absorbance spectra are
generated from these data. (If necessary, apply the spectral transformations
developed in section 5.6.2 of this addendum). The resulting sample spectrum is
referred to below as SS.
Note: Multiple sample
spectra may be recorded according to the procedures of section 5.4 of this
addendum before performing sections 5.5 and 5.6 of this addendum.
5.5 Quantify Analyte Concentrations.
Calculate the unscaled
analyte concentrations RUAi
and unscaled interferant concentrations
RUIK using the programs developed in section 4 of this
addendum. To correct for pathlength and pressure variations between the
reference and sample spectra, calculate the scaling factor, RLPS using equation A.1,
![]()
Calculate the final
analyte and interferant concentrations RSAi and RSIk using equations A.2 and A.3,

5.6 Determine Fractional Analysis Uncertainty.
Fill the absorption cell
with CTS at the pressure PS. Record a set of CTS spectra {R4}. Store the
background and CTS single beam interferograms. Using appendix H of this
addendum, calculate the fractional analysis uncertainty (FAU) for each
analytical region. If the FAU indicated for any analytical region is greater
than the required accuracy requirements determined in sections 4.1.1 through
4.1.4 of this addendum, then comparisons to previously recorded reference
spectra are invalid in that analytical region, and the analyst shall perform
one or both of the procedures of sections 5.6.1 through 5.6.2 of this addendum.
5.6.1 Perform instrumental
checks and adjust the instrument to restore its performance to acceptable
levels. If adjustments are made, repeat sections 5.3, 5.4 (except for the
recording of a sample spectrum), and 5.5 of this addendum to demonstrate that
acceptable uncertainties are obtained in all analytical regions.
5.6.2 Apply appropriate
mathematical transformations (e.g., frequency shifting, zero-filling,
apodization, smoothing) to the spectra (or to the interferograms upon which the
spectra are based) generated during the performance of the procedures of
section 5.3 of this addendum. Document these transformations and their
reproducibility. Do not apply multiplicative scaling of the spectra, or any set
of transformations that is mathematically equivalent to multiplicative scaling.
Different transformations may be applied to different analytical regions.
Frequency shifts shall be less than one-half the minimum instrumental
line-width, and must be applied to all spectral data points in an analytical
region. The mathematical transformations may be retained for the analysis if
they are also applied to the appropriate analytical regions of all sample
spectra recorded, and if all original sample spectra are digitally stored.
Repeat sections 5.3, 5.4 (except the recording of a sample spectrum), and 5.5
of this addendum to demonstrate that these transformations lead to acceptable
calculated concentration uncertainties in all analytical regions.
6.0 POST-ANALYSIS EVALUATIONS
Estimate the overall
accuracy of the analyses performed in accordance with sections 5.1 through 5.6
of this addendum using the procedures of sections 6.1 through 6.3 of this
addendum.
6.1 Qualitatively Confirm the Assumed Matrix.
Examine each analytical
region of the sample spectrum for spectral evidence of unexpected or unidentified
interferants. If found, identify the interfering compounds (see Reference C for
guidance) and add them to the list of known interferants. Repeat the procedures
of section 4 of this addendum to include the interferants in the uncertainty
calculations and analysis procedures. Verify that the MAU and FCU values do not
increase beyond acceptable levels for the application requirements.
Re-calculate the analyte concentrations (section 5.5 of this addendum) in the
affected analytical regions.
6.2 Quantitatively Evaluate Fractional Model Uncertainty (FMU).
Perform the procedures of
either section 6.2.1 or 6.2.2 of this addendum:
6.2.1 Using appendix I of
this addendum, determine the fractional model error (FMU) for each analyte.
6.2.2 Provide
statistically determined uncertainties FMU for each analyte which are
equivalent to two standard deviations at the 95 percent confidence level. Such
determinations, if employed, must be based on mathematical examinations of the
pertinent sample spectra (not the reference spectra alone). Include in the
report of the analysis (see section 7.0 of this addendum) a complete
description of the determination of the concentration uncertainties.
6.3 Estimate Overall Concentration Uncertainty (OCU).
Using appendix J of this
addendum, determine the overall concentration uncertainty (OCU) for each
analyte. If the OCU is larger than the required accuracy for any analyte,
repeat sections 4 and 6 of this addendum.
7.0 REPORTING REQUIREMENTS
[Documentation
pertaining to virtually all the procedures of sections 4, 5, and 6 will be
required. Software copies of reference spectra and sample spectra will be
retained for some minimum time following the actual testing.]
8.0 REFERENCES
A) Standard Practices for
General Techniques of Infrared Quantitative Analysis (American Society for
Testing and Materials, Designation E 168-88).
B) The Coblentz Society
Specifications for Evaluation of Research Quality Analytical Infrared Reference
Spectra (Class II); Anal. Chemistry 47, 945A (1975); Appl. Spectroscopy 444,
pp. 211-215, 1990.
C) Standard Practices for
General Techniques for Qualitative Infrared Analysis, American Society for
Testing and Materials, Designation E 1252-88.
D) "EPA Traceability
Protocol for Assay and Certification of Gaseous Calibration Standards,"
U.S. Environmental Protection Agency Publication No. EPA/600/R-93/224, December
1993.
APPENDIX A to Addendum to Method 320
DEFINITIONS OF TERMS AND
SYMBOLS
A.1 Definitions of Terms.
All terms used in this
method that are not defined below have the meaning given to them in the CAA and
in subpart A of this part.
Absorption band means a contiguous wavenumber region of a spectrum
(equivalently, a contiguous set of absorbance spectrum data points) in which
the absorbance passes through a maximum or a series of maxima.
Absorption
pathlength means the distance in
a spectrophotometer, measured in the direction of propagation of the beam of
radiant energy, between the surface of the specimen on which the radiant energy
is incident and the surface of the specimen from which it is emergent.
Analytical region means a contiguous wavenumber region (equivalently,
a contiguous set of absorbance spectrum data points) used in the quantitative
analysis for one or more analytes.
Note: The quantitative
result for a single analyte may be based on data from more than one analytical
region. Apodization means modification of the ILS function by multiplying the
interferogram by a weighing function whose magnitude varies with retardation.
Background spectrum means the single beam spectrum obtained with all
system components without sample present.
Baseline means any line drawn on an absorption spectrum to
establish a reference point that represents a function of the radiant power
incident on a sample at a given wavelength.
Beers's law means the direct proportionality of the absorbance
of a compound in a homogeneous sample to its concentration.
Calibration transfer
standard (CTS) gas means a gas
standard of a compound used to achieve and/or demonstrate suitable quantitative
agreement between sample spectra and the reference spectra; see section 4.5.1
of this addendum.
Compound means a substance possessing a distinct, unique
molecular structure.
Concentration (c) means the quantity of a compound contained in a unit
quantity of sample. The unit "ppm" (number, or mole, basis) is
recommended.
Concentration-pathlength
product means the mathematical
product of concentration of the species and absorption pathlength. For
reference spectra, this is a known quantity; for sample spectra, it is the
quantity directly determined from Beer's law. The units
"centimeters-ppm" or "meters-ppm" are recommended.
Derivative
absorption spectrum means a plot
of rate of change of absorbance or of any function of absorbance with respect
to wavelength or any function of wavelength.
Double beam spectrum
means a transmission or
absorbance spectrum derived by dividing the sample single beam spectrum by the
background spectrum.
Note: The term
"double-beam" is used elsewhere to denote a spectrum in which the
sample and background interferograms are collected simultaneously along
physically distinct absorption paths. Here, the term denotes a spectrum in
which the sample and background interferograms are collected at different times
along the same absorption path.
Fast Fourier
transform (FFT) means a method
of speeding up the computation of a discrete FT by factoring the data into
sparse matrices containing mostly zeros.
Flyback means interferometer motion during which no data are
recorded.
Fourier transform
(FT) means the mathematical
process for converting an amplitude-time spectrum to an amplitude frequency
spectrum, or vice versa.
Fourier transform
infrared (FTIR) spectrometer
means an analytical system that employs a source of mid-infrared radiation, an
interferometer, an enclosed sample cell of known absorption pathlength, an
infrared detector, optical elements that transfer infrared radiation between
components, and a computer system. The time-domain detector response (interferogram)
is processed by a Fourier transform to yield a representation of the detector
response vs. infrared frequency.
Note: When FTIR
spectrometers are interfaced with other instruments, a slash should be used to
denote the interface; e.g., GC/FTIR; HPCL/FTIR, and the use of FTIR should be
explicit; i.e., FTIR not IR.
Frequency, v
means the number of cycles per
unit time.
Infrared means the portion of the electromagnetic spectrum
containing wavelengths from approximately 0.78 to 800 microns.
Interferogram, I(•) means record of the modulated component of the
interference signal measured as a function of retardation by the detector.
Interferometer means device that divides a beam of radiant energy
into two or more paths, generates an optical path difference between the beams,
and recombines them in order to produce repetitive interference maxima and
minima as the optical retardation is varied.
Linewidth means the full width at half maximum of an
absorption band in units of wavenumbers (cm-1). Mid-infrared
means the region of the electromagnetic spectrum from approximately 400 to 5000
cm-1.
Reference spectra means absorption spectra of gases with known
chemical compositions, recorded at a known absorption pathlength, which are
used in the quantitative analysis of gas samples.
Retardation, • means optical path difference between two beams in
an interferometer; also known as "optical path difference" or
"optical retardation."
Scan means digital representation of the detector output
obtained during one complete motion of the interferometer's moving assembly or
assemblies.
Scaling means application of a multiplicative factor to the
absorbance values in a spectrum.
Single beam spectrum means Fourier-transformed interferogram,
representing the detector response vs. wavenumber.
Note: The term
"single-beam" is used elsewhere to denote any spectrum in which the
sample and background interferograms are recorded on the same physical
absorption path; such usage differentiates such spectra from those generated
using interferograms recorded along two physically distinct absorption paths
(see "double-beam spectrum" above). Here, the term applies (for
example) to the two spectra used directly in the calculation of transmission
and absorbance spectra of a sample.
Standard reference
material means a reference
material, the composition or properties of which are certified by a recognized
standardizing agency or group.
Note: The equivalent ISO
term is "certified reference material."
Transmittance, T means
the ratio of radiant power transmitted by the sample to the radiant power
incident on the sample. Estimated in FTIR spectroscopy by forming the ratio of
the single-beam sample and background spectra.
Wavenumber, v means
the number of waves per unit length.
Note: The usual unit of
wavenumber is the reciprocal centimeter, cm-1. The wavenumber is
the reciprocal of the wavelength, 8, when 8 is expressed in centimeters.
Zero-filling means the addition of zero-valued points to the end
of a measured interferogram.
Note: Performing the FT of
a zero-filled interferogram results in correctly interpolated points in the
computed spectrum.
A.2 Definitions of Mathematical Symbols.
The symbols used in
equations in this protocol are defined as follows:
(1) A, absorbance = the
logarithm to the base 10 of the reciprocal of the transmittance (T).
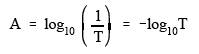
(2) AAIim = band area of the ith analyte in the mth analytical region, at the concentration (CLi) corresponding to the product of its required detection limit (DLi) and analytical uncertainty limit (AUi) .
(3) AAVim = average absorbance of the ith analyte in the mth analytical region, at the concentration (CLi) corresponding to the product of its required detection limit (DLi) and analytical uncertainty limit (AUi) .
(4) ASC, accepted standard
concentration = the concentration value assigned to a chemical standard.
(5) ASCPP, accepted
standard concentration-pathlength product = for a chemical standard, the
product of the ASC and the sample absorption pathlength. The units
"centimeters-ppm" or "meters-ppm" are recommended.
(6) AUi, analytical uncertainty limit = the maximum permissible fractional
uncertainty of analysis for the ith analyte
concentration, expressed as a fraction of the analyte concentration determined
in the analysis.
(7) AVTm = average estimated total absorbance in the mth analytical region.
(8) CKWNk = estimated concentration of the kth known interferant.
(9) CMAXi = estimated maximum concentration of the ith analyte.
(10) CPOTj = estimated concentration of the jth potential
interferant.
(11) DLi, required detection limit = for the ith analyte, the
lowest concentration of the analyte for which its overall fractional
uncertainty (OFUi) is required to be less than the analytical
uncertainty limit (AUi).
(12) FCm = center wavenumber position of the mth analytical region.
(13) FAUi, fractional analytical uncertainty = calculated uncertainty in the
measured concentration of the ith analyte because of errors in
the mathematical comparison of reference and sample spectra.
(14) FCUi, fractional calibration uncertainty = calculated uncertainty in the
measured concentration of the ith analyte because of errors in
Beer's law modeling of the reference spectra concentrations.
(15) FFLm = lower wavenumber position of the CTS absorption band associated with
the mth analytical region.
(16) FFUm = upper wavenumber position of the CTS absorption band associated with
the mth analytical region.
(17) FLm = lower wavenumber position of the mth analytical region.
(18) FMUi, fractional model uncertainty = calculated uncertainty in the measured
concentration of the ith
analyte because of errors in the
absorption model employed.
(19) FNL = lower wavenumber position of the CTS spectrum containing an absorption
band at least as narrow as the analyte absorption bands.
(20) FNU = upper wavenumber position of the CTS spectrum containing an absorption
band at least as narrow as the analyte absorption bands.
(21) FRUi, fractional reproducibility uncertainty = calculated uncertainty in the
measured concentration of the ith analyte because of errors in
the reproducibility of spectra from the FTIR system.
(22) FUm = upper wavenumber position of the mth analytical region.
(23) IAIjm = band area of the jth potential interferant in the mth analytical region, at its expected concentration (CPOTj).
(24) IAVim = average absorbance of the ith analyte in the mth analytical region, at its expected concentration (CPOTj).
(25) ISCi or k, indicated standard concentration = the concentration from the
computerized analytical program for a single-compound reference spectrum for
the ith analyte or kth known interferant.
(26) kPa = kilo-Pascal
(see Pascal).
(27) LS' = estimated sample absorption pathlength.
(28) LR = reference absorption pathlength.
(29) LS = actual sample absorption pathlength.
(30) MAUi = mean of the MAUim
over the appropriate analytical regions.
(31) MAUim, minimum analyte uncertainty = the calculated minimum concentration for
which the analytical uncertainty limit (AUi) in the measurement
of the ith analyte, based on spectral data in the mth analytical region, can be maintained.
(32) MIUj = mean of the MIUjm
over the appropriate analytical regions.
(33) MIUjm, minimum interferant uncertainty = the calculated minimum concentration
for which the analytical uncertainty limit CPOTj/20 in the
measurement of the jth interferant, based on spectral data in the mth analytical region, can be maintained.
(34) MIL, minimum
instrumental line-width = the minimum line-width from the FTIR system, in wavenumbers.
Note: The MIL of a system
may be determined by observing an absorption band known (through higher
resolution examinations) to be narrower than indicated by the system. The MIL
is fundamentally limited by the retardation of the interferometer, but is also
affected by other operational parameters (e.g., the choice of apodization).
(35) Ni = number of analytes.
(36) Nj = number of potential interferants.
(37) Nk = number of known interferants.
(38) Nscan = the number of scans averaged to obtain an
interferogram.
(39) OFUi = the overall fractional uncertainty in an analyte concentration
determined in the analysis (OFUi = MAX{FRUi, FCUi, FAUi, FMUi}).
(40) Pascal (Pa) = metric
unit of static pressure, equal to one Newton per square meter; one atmosphere
is equal to 101,325 Pa; 1/760 atmosphere (one Torr, or one millimeter Hg) is
equal to 133.322 Pa.
(41) Pmin = minimum pressure of the sampling system during the sampling procedure.
(42) PS' = estimated sample pressure.
(43) PR = reference pressure.
(44) PS = actual sample pressure.
(45) RMSSm = measured noise level of the FTIR system in the mth analytical region.
(46) RMSD, root mean
square difference = a measure of accuracy determined by the following equation:
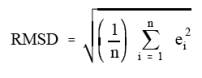
where:

Note: The RMSD value
"between a set of n contiguous absorbance values (Ai) and the mean of the values" (AM) is defined as
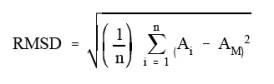
(47) RSAi = the (calculated) final concentration of the ith analyte.
(48) RSIk = the (calculated) final concentration of the kth known interferant.
(49) tscan, scan time = time used to acquire a single scan, not including flyback.
(50) tS, signal integration period = the period of time over which an
interferogram is averaged by addition and scaling of individual scans. In terms
of the number of scans Nscan
and scan time tscan, tS = Nscantscan.
(51) tSR = signal integration period used in recording reference spectra.
(52) tSS = signal integration period used in recording sample spectra.
(53) TR = absolute temperature of gases used in recording reference spectra.
(54) TS = absolute temperature of sample gas as sample spectra are recorded.
(55) TP, Throughput =
manufacturer's estimate of the fraction of the total infrared power transmitted
by the absorption cell and transfer optics from the interferometer to the
detector.
(56) VSS = volume of the infrared absorption cell, including parts of attached
tubing.
(57) Wik = weight used to average over analytical regions k for quantities
related to the analyte i; see appendix D of this addendum.
APPENDIX B TO ADDENDUM TO METHOD 320
IDENTIFYING SPECTRAL
INTERFERANTS
B.1 General
B.1.1 Assume a fixed
absorption pathlength equal to the value LS'.
B.1.2 Use band area
calculations to compare the relative absorption strengths of the analytes and
potential interferants. In the mth analytical region (FLm to FUm), use either rectangular or trapezoidal
approximations to determine the band areas described below (see Reference A,
sections A.3.1 through A.3.3). Document any baseline corrections applied to the
spectra.
B.1.3 Use the average
total absorbance of the analytes and potential interferants in each analytical
region to determine whether the analytical region is suitable for analyte
concentration determinations.
Note: The average
absorbance in an analytical region is the band area divided by the width of the
analytical region in wavenumbers. The average total absorbance in an analytical
region is the sum of the average absorbances of all analytes and potential
interferants.
B.2 Calculations
B.2.1 Prepare spectral
representations of each analyte at the concentration CLi = (DLi)(AUi), where DLi is the required detection limit and AUi is the
maximum permissible analytical uncertainty. For the mth analytical region, calculate the band area (AAIim) and average absorbance (AAVim) from these scaled
analyte spectra.
B.2.2 Prepare spectral
representations of each potential interferant at its expected concentration
(CPOTj). For the mth analytical region,
calculate the band area (IAIjm) and average absorbance (IAVjm) from these scaled potential interferant spectra.
B.2.3 Repeat the
calculation for each analytical region, and record the band area results in
matrix form as indicated in Figure B.1.
B.2.4 If the band area of
any potential interferant in an analytical region is greater than the one-half
the band area of any analyte (i.e., IAIjm > 0.5 AAIim for any pair ij and any m), classify the potential interferant as a
known interferant. Label the known interferants k = 1 to K. Record the results
in matrix form as indicated in Figure B.2.
B.2.5 Calculate the
average total absorbance (AVTm) for each analytical region and
record the values in the last row of the matrix described in Figure B.2. Any
analytical region where AVTm >2.0 is unsuitable.
FIGURE B.1 Presentation of Potential Interferant Calculations.

FIGURE B.2 Presentation of Known Interferant Calculations

APPENDIX C TO ADDENDUM TO METHOD 320
ESTIMATING NOISE LEVELS
C.1 General
C.1.1 The root-mean-square
(RMS) noise level is the standard measure of noise in this addendum. The RMS
noise level of a contiguous segment of a spectrum is defined as the RMS
difference (RMSD) between the absorbance values which form the segment and the
mean value of that segment (see appendix A of this addendum).
C.1.2 The RMS noise value
in double-beam absorbance spectra is assumed to be inversely proportional to:
(a) the square root of the signal integration period of the sample single beam
spectra from which it is formed, and (b) the total infrared power transmitted
through the interferometer and absorption cell.
C.1.3 Practically, the
assumption of C.1.2 allows the RMS noise level of a complete system to be
estimated from the quantities described in sections C.1.3.1 through C.1.3.4:
C.1.3.1 RMSMAN, the noise level of the system (in absorbance units), without the
absorption cell and transfer optics, under those conditions necessary to
yield the specified minimum instrumental line-width, e.g., Jacquinot stop
size.
C.1.3.2 tMAN, the manufacturer's signal integration time used to determine RMSMAN.
C.1.3.3 tSS, the signal integration time for the analyses.
C.1.3.4 TP, the
manufacturer's estimate of the fraction of the total infrared power transmitted
by the absorption cell and transfer optics from the interferometer to the
detector.
C.2 Calculations
C.2.1 Obtain the values of
RMSMAN, tMAN, and TP from the
manufacturers of the equipment, or determine the noise level by direct
measurements with the completely constructed system proposed in section 4 of
this addendum.
C.2.2 Calculate the noise
value of the system (RMSEST) using equation C.1.
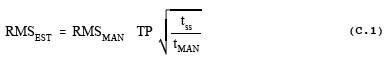
APPENDIX D TO ADDENDUM TO METHOD 320
ESTIMATING MINIMUM
CONCENTRATION MEASUREMENT UNCERTAINTIES (MAU and MIU)
D.1 General
Estimate the minimum
concentration measurement uncertainties for the ith analyte (MAUi) and jth interferant (MIUj) based on the
spectral data in the mth
analytical region by comparing the
analyte band area in the analytical region (AAIim) and
estimating or measuring the noise level of the system (RMSEST or RMSSm).
Note: For a single
analytical region, the MAU or MIU value is the concentration of the analyte or
interferant for which the band area is equal to the product of the analytical
region width (in wavenumbers) and the noise level of the system (in absorbance
units). If data from more than one analytical region are used in the
determination of an analyte concentration, the MAU or MIU is the mean of the
separate MAU or MIU values calculated for each analytical region.
D.2 Calculations
D.2.1 For each analytical
region, set RMS = RMSSm if measured (appendix G of this addendum), or set RMS
= RMSEST if estimated (appendix C of this addendum).
D.2.2 For each analyte
associated with the analytical region, calculate MAUim using equation D.1,
D.2.3 If only the mth analytical region is used to calculate the concentration of the ith analyte, set MAUi
= MAUim.
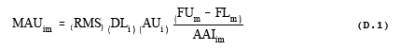
D.2.4 If more than one
analytical region is used to calculate the concentration of the ith analyte, set MAUi
equal to the weighted mean of the appropriate
MAUim values calculated above; the weight for each term in
the mean is equal to the fraction of the total wavenumber range used for the
calculation represented by each analytical region. Mathematically, if the set
of analytical regions employed is {m'}, then the MAU for each analytical region
is given by equation D.2.
![]()
where the weight Wik is defined for each term in the sum as
![]()
D.2.5 Repeat sections
D.2.1 through D.2.4 of this appendix to calculate the analogous values MIUj for the interferants j = 1 to J. Replace the value (AUi)(DLi) in equation D.1 with CPOTj/20; replace the value AAIim in equation D.1
with IAIjm.
APPENDIX E TO ADDENDUM TO METHOD 320
DETERMINING FRACTIONAL
REPRODUCIBILITY UNCERTAINTIES (FRU)
E.1 General
To estimate the
reproducibility of the spectroscopic results of the system, compare the CTS
spectra recorded before and after preparing the reference spectra. Compare the
difference between the spectra to their average band area. Perform the
calculation for each analytical region on the portions of the CTS spectra
associated with that analytical region.
E.2 Calculations
E.2.1 The CTS spectra {R1}
consist of N spectra, denoted by S1i, i=1, N.
Similarly, the CTS spectra {R2} consist of N spectra, denoted by S2i, i=1, N. Each Ski
is the spectrum of a single compound,
where i denotes the compound and k denotes the set {Rk} of which Ski is a member. Form the spectra S3 according to S3i = S2i-S1i for each i. Form the spectra S4 according to S4i = [S2i+S1i]/2 for each i.
E.2.2 Each analytical
region m is associated with a portion of the CTS spectra S2i and S1i, for a particular i, with lower and upper wavenumber
limits FFLm and FFUm, respectively.
E.2.3 For each m and the
associated i, calculate the band area of S4i in the wavenumber
range FFUm to FFLm. Follow the guidelines of
section B.1.2 of this addendum for this band area calculation. Denote the
result by BAVm.
E.2.4 For each m and the
associated i, calculate the RMSD of S3i between the
absorbance values and their mean in the wavenumber range FFUm to FFLm. Denote the result by SRMSm.
E.2.5 For each analytical
region m, calculate FMm using equation E.1,
![]()
E.2.6 If only the mth analytical region is used to calculate the concentration of the ith analyte, set FRUi
= FMm.
E.2.7 If a number pi of analytical regions are used to calculate the concentration of the ith analyte, set FRUi
equal to the weighted mean of the
appropriate FMm values calculated according to section E.2.5.
Mathematically, if the set of analytical regions employed is {m'}, then FRUi is given by equation E.2,
![]()
where the Wik are calculated as described in appendix D of this addendum.
APPENDIX F OF ADDENDUM TO METHOD 320
DETERMINING FRACTIONAL
CALIBRATION UNCERTAINTIES (FCU)
F.1 General
F.1.1 The concentrations
yielded by the computerized analytical program applied to each single-compound
reference spectrum are defined as the indicated standard concentrations
(ISC's). The ISC values for a single compound spectrum should ideally equal the
accepted standard concentration (ASC) for one analyte or interferant, and
should ideally be zero for all other compounds. Variations from these results
are caused by errors in the ASC values, variations from the Beer's law (or
modified Beer's law) model used to determine the concentrations, and noise in
the spectra. When the first two effects dominate, the systematic nature of the
errors is often apparent and the analyst shall take steps to correct them.
F.1.2 When the calibration
error appears nonsystematic, apply the procedures of sections F.2.1 through
F.2.3 of this appendix to estimate the fractional calibration uncertainty (FCU)
for each compound. The FCU is defined as the mean fractional error between the
ASC and the ISC for all reference spectra with non-zero ASC for that compound.
The FCU for each compound shall be less than the required fractional
uncertainty specified in section 4.1 of this addendum.
F.1.3 The computerized
analytical programs shall also be required to yield acceptably low
concentrations for compounds with ISC = 0 when applied to the reference
spectra. The ISC of each reference spectrum for each analyte or interferant
shall not exceed that compound's minimum measurement uncertainty (MAU or MIU).
F.2 Calculations
F.2.1 Apply each
analytical program to each reference spectrum. Prepare a similar table to that
in Figure F.1 to present the ISC and ASC values for each analyte and
interferant in each reference spectrum. Maintain the order of reference file
names and compounds employed in preparing Figure F.1.
F.2.2 For all reference
spectra in Figure F.1, verify that the absolute values of the ISC's are less
than the compound's MAU (for analytes) or MIU (for interferants).
F.2.3 For each analyte
reference spectrum, calculate the quantity (ASC-ISC)/ASC. For each analyte,
calculate the mean of these values (the FCUi for the ith analyte) over all reference spectra. Prepare a similar table to that in
Figure F.2 to present the FCUi and analytical uncertainty
limit (AUi) for each analyte.
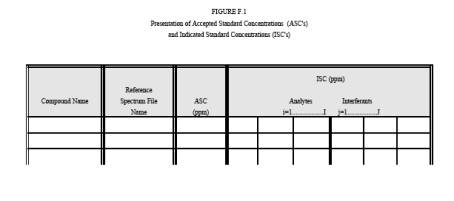

APPENDIX G TO ADDENDUM TO METHOD 320
MEASURING NOISE LEVELS
G.1 General
The root-mean-square (RMS)
noise level is the standard measure of noise. The RMS noise level of a
contiguous segment of a spectrum is the RMSD between the absorbance values that
form the segment and the mean value of the segment (see appendix A of this
addendum).
G.2 Calculations
G.2.1 Evacuate the
absorption cell or fill it with UPC grade nitrogen at approximately one
atmosphere total pressure.
G.2.2 Record two single
beam spectra of signal integration period tSS.
G.2.3 Form the double beam
absorption spectrum from these two single beam spectra, and calculate the noise
level RMSSm in the M analytical regions.
APPENDIX H OF ADDENDUM TO METHOD 320
DETERMINING SAMPLE
ABSORPTION PATHLENGTH (LS) AND FRACTIONAL ANALYTICAL UNCERTAINTY (FAU)
H.1 General
Reference spectra recorded
at absorption pathlength (LR), gas pressure (PR), and gas absolute temperature (TR) may be used to
determine analyte concentrations in samples whose spectra are recorded at
conditions different from that of the reference spectra, i.e., at absorption
pathlength (LS), absolute temperature (TS), and pressure (PS). This appendix describes the calculations for
estimating the fractional uncertainty (FAU) of this practice. It also describes
the calculations for determining the sample absorption pathlength from
comparison of CTS spectra, and for preparing spectra for further instrumental
and procedural checks.
H.1.1 Before sampling,
determine the sample absorption pathlength using least squares analysis.
Determine the ratio LS/LR by comparing the spectral sets
{R1} and {R3}, which are recorded using the same CTS at LS and LR, and TS and TR, but both at PR.
H.1.2 Determine the
fractional analysis uncertainty (FAU) for each analyte by comparing a scaled
CTS spectral set, recorded at LS, TS, and PS, to the CTS reference spectra of the same gas,
recorded at LR, TR, and PR. Perform the quantitative comparison after recording the sample
spectra, based on band areas of the spectra in the CTS absorbance band
associated with each analyte.
H.2 Calculations
H.2.1 Absorption
Pathlength Determination. Perform and document separate linear baseline
corrections to each analytical region in the spectral sets {R1} and {R3}. Form
a one-dimensional array AR containing the absorbance
values from all segments of {R1} that are associated with the analytical
regions; the members of the array are ARi, i = 1, n. Form a
similar one-dimensional array AS from the absorbance
values in the spectral set {R3}; the members of the array are ASi, i = 1, n. Based on the model AS = rAR + E, determine the
least-squares estimate of r', the value of r which minimizes the square error E2. Calculate the sample absorption pathlength, LS, using equation H.1,
![]()
H.2.2 Fractional Analysis
Uncertainty. Perform and document separate linear baseline corrections to each
analytical region in the spectral sets {R1} and {R4}. Form the arrays AS and AR as described in section H.2.1 of this appendix, using
values from {R1} to form AR, and values from
{R4} to form AS. Calculate NRMSE and IAAV using equations H.2 and H.3,

The fractional analytical
uncertainty, FAU, is given by equation H.4,

APPENDIX I TO ADDENDUM TO METHOD 320
DETERMINING FRACTIONAL
MODEL UNCERTAINTIES (FMU)
I.1 General
To prepare analytical
programs for FTIR analyses, the sample constituents must first be assumed. The
calculations in this appendix, based upon a simulation of the sample spectrum,
shall be used to verify the appropriateness of these assumptions. The simulated
spectra consist of the sum of single compound reference spectra scaled to
represent their contributions to the sample absorbance spectrum; scaling
factors are based on the indicated standard concentrations (ISC) and measured
(sample) analyte and interferant concentrations, the sample and reference
absorption pathlengths, and the sample and reference gas pressures. No
band-shape correction for differences in the temperature of the sample and
reference spectra gases is made; such errors are included in the FMU estimate.
The actual and simulated sample spectra are quantitatively compared to
determine the fractional model uncertainty; this comparison uses the reference
spectra band areas and residuals in the difference spectrum formed from the actual
and simulated sample spectra.
I.2 Calculations
I.2.1 For each analyte
(with scaled concentration RSAi), select a reference spectrum
SAi with indicated standard concentration ISCi. Calculate the scaling factors, RAi, using equation
I.1,

Form the spectra SACi by scaling each SAi
by the factor RAi.
I.2.2 For each
interferant, select a reference spectrum SIk with indicated
standard concentration ISCk. Calculate the scaling factors, RIk, using equation I.2,

Form the spectra SICk by scaling each SIk
by the factor RIk.
I.2.3 For each analytical
region, determine by visual inspection which of the spectra SACi and SICk exhibit absorbance bands within the analytical
region. Subtract each spectrum SACi and SICk exhibiting absorbance from the sample spectrum SS to form the spectrum SUBS. To save analysis time and to
avoid the introduction of unwanted noise into the subtracted spectrum, it is
recommended that the calculation be made (1) only for those spectral data
points within the analytical regions, and (2) for each analytical region
separately using the original spectrum SS.
I.2.4 For each analytical
region m, calculate the RMSD of SUBS between the
absorbance values and their mean in the region FFUm to FFLm. Denote the result by RMSSm.
I.2.5 For each analyte i,
calculate FMm, using equation I.3,

for each analytical region
associated with the analyte.
I.2.6 If only the mth analytical region is used to calculate the concentration of the ith analyte, set FMUi=FMm.
I.2.7 If a number of
analytical regions are used to calculate the concentration of the ith analyte, set FMi equal to the weighted mean of the appropriate FMm values calculated using equation I-3. Mathematically, if the set of
analytical regions employed is {m'}, then the fractional model uncertainty,
FMU, is given by equation I.4,
![]()
where Wik is calculated as described in appendix D of this addendum.
APPENDIX J OF ADDENDUM TO METHOD 320
DETERMINING OVERALL
CONCENTRATION UNCERTAINTIES (OCU)
The calculations in this
addendum estimate the measurement uncertainties for various FTIR measurements.
The lowest possible overall concentration uncertainty (OCU) for an analyte is
its MAU value, which is an estimate of the absolute concentration uncertainty
when spectral noise dominates the measurement error. However, if the product of
the largest fractional concentration uncertainty (FRU, FCU, FAU, or FMU) and
the measured concentration of an analyte exceeds the MAU for the analyte, then
the OCU is this product. In mathematical terms, set OFUi = MAX{FRUi, FCUi, FAUi, FMUi} and OCUi = MAX{RSAi*OFUi, MAUi}.
Figure 2. FTIR
Sampling/Spiking System.