METHOD 15 -
DETERMINATION OF HYDROGEN SULFIDE, CARBONYL SULFIDE, AND CARBON DISULFIDE EMISSIONS
FROM STATIONARY SOURCES
NOTE: This method is not inclusive with respect to
specifications (e.g.,
equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance.
Some material is incorporated by reference from other methods in this part.
Therefore, to obtain reliable results, persons using this method should have a
thorough knowledge of gas chromatography techniques.
4.2 Carbon Monoxide
(CO) and Carbon Dioxide (CO2).
6.3.2 Constant
Temperature Bath.
8.0 Sample Collection,
Preservation, Transport, and Storage.
8.2 Sample Collection
and Analysis.
8.3.2 Determination of
Calibration Drift.
10.0 Calibration and
Standardization.
10.1 Calibration Gas
Permeation Tube Preparation.
10.2 Calibration of
Analytical System.
10.4 Calibration of
Dilution System.
12.0 Data Analysis and
Calculations.
14.0 Pollution
Prevention. [Reserved]
15.0 Waste Management.
[Reserved]
17.0 Tables, Diagrams,
Flowcharts, and Validation Data.
1.0 Scope and Application.
1.1 Analytes.

1.2 Applicability.
1.2.1 This method
applies to the determination of emissions of reduced sulfur compounds from tail
gas control units of sulfur recovery plants, H2S in
fuel gas for fuel gas combustion devices, and where specified in other applicable
subparts of the regulations.
1.2.2 The method
described below uses the principle of gas chromatographic (GC) separation and
flame photometric detection (FPD). Since there are many systems or sets of
operating conditions that represent useable methods for determining sulfur
emissions, all systems which employ this principle, but differ only in details
of equipment and operation, may be used as alternative methods, provided that
the calibration precision and sample-line loss criteria are met.
1.3 Data Quality Objectives.
Adherence to the
requirements of this method will enhance the quality of the data obtained from
air pollutant sampling methods.
2.0 Summary of Method.
2.1 A gas sample is
extracted from the emission source and diluted with clean dry air (if
necessary). An aliquot of the diluted sample is then analyzed for CS2, COS, and H2S by GC/FPD.
3.0 Definitions. [Reserved]
4.0 Interferences.
4.1 Moisture Condensation.
Moisture condensation
in the sample delivery system, the analytical column, or the FPD burner block
can cause losses or interferences. This potential is eliminated by heating the
probe, filter box, and connections, and by maintaining the SO2 scrubber in an ice water bath. Moisture is removed in the SO2 scrubber and heating the sample beyond this point is not necessary
provided the ambient temperature is above 0 ˚C (32 ˚F). Alternatively, moisture
may be eliminated by heating the sample line, and by conditioning the sample
with dry dilution air to lower its dew point below the operating temperature of
the GC/FPD analytical system prior to analysis.
4.2 Carbon Monoxide (CO) and Carbon Dioxide (CO2).
CO and CO2 have substantial desensitizing effects on the FPD even after 9:1
dilution. (Acceptable systems must demonstrate that they have eliminated this
interference by some procedure such as eluting CO and CO2 before any of the sulfur compounds to be measured.) Compliance with
this requirement can be demonstrated by submitting chromatograms of calibration
gases with and without CO2
in the diluent gas. The CO2 level should be approximately 10 percent for the case with CO2 present. The two chromatograms should show agreement within the
precision limits of Section 13.3.
4.3 Elemental Sulfur.
The condensation of
sulfur vapor in the sampling system can lead to blockage of the particulate
filter. This problem can be minimized by observing the filter for buildup and
changing as needed.
4.4 Sulfur Dioxide (SO2).
SO2 is not a specific interferent but may be present in such large
amounts that it cannot be effectively separated from the other compounds of
interest. The SO2 scrubber described in Section 6.1.3 will
effectively remove SO2
from the sample.
4.5 Alkali Mist.
Alkali mist in the
emissions of some control devices may cause a rapid increase in the SO2 scrubber pH, resulting in low sample recoveries. Replacing the SO2 scrubber contents after each run will minimize the chances of
interference in these cases.
5.0 Safety.
5.1 Disclaimer. This
method may involve hazardous materials, operations, and equipment. This test
method may not address all of the safety problems associated with its use. It
is the responsibility of the user of this test to establish appropriate safety
and health practices and determine the applicability of regulatory limitations
to performing this test.
6.0 Equipment and Supplies.
6.1 Sample Collection.
See Figure 15-1. The
sampling train component parts are discussed in the following sections:
6.1.1 Probe.
The probe shall be
made of Teflon or Teflon-lined stainless steel and heated to prevent moisture
condensation. It shall be designed to allow calibration gas to enter the probe
at or near the sample point entry. Any portion of the probe that contacts the
stack gas must be heated to prevent moisture condensation. The probe described
in Section 6.1.1 of Method 16A having a nozzle directed away from the gas
stream is recommended for sources having particulate or mist emissions. Where
very high stack temperatures prohibit the use of Teflon probe components, glass
or quartz-lined probes may serve as substitutes.
6.1.2 Particulate Filter.
50-mm Teflon filter
holder and a 1-to 2-micron porosity Teflon filter (available through Savillex
Corporation, 5325 Highway 101, Minnetonka, Minnesota 55343). The filter holder
must be maintained in a hot box at a temperature of at least 120 ˚C (248 ˚F).
6.1.3 SO2 Scrubber.
Three 300-ml Teflon
segment impingers connected in series with flexible, thick-walled, Teflon
tubing. (Impinger parts and tubing available through Savillex.) The first two
impingers contain 100 ml of citrate buffer, and the third impinger is initially
dry. The tip of the tube inserted into the solution should be constricted to
less than 3-mm (c-in.) ID and should be immersed to a depth of at least 50 cm
(2 in.). Immerse the impingers in an ice water bath and maintain near 0 ˚C. The
scrubber solution will normally last for a 3-hour run before needing
replacement. This will depend upon the effects of moisture and particulate
matter on the solution strength and pH. Connections between the probe,
particulate filter, and SO2
scrubber shall be made of Teflon
and as short in length as possible. All portions of the probe, particulate
filter, and connections prior to the SO2 scrubber
(or alternative point of moisture removal) shall be maintained at a temperature
of at least 120 ˚C (248 ˚F).
6.1.4 Sample Line.
Teflon, no greater
than 13-mm (1/2-in.) ID. Alternative materials, such as virgin Nylon, may be
used provided the line-loss test is acceptable.
6.1.5 Sample Pump.
The sample pump shall
be a leakless Teflon-coated diaphragm type or equivalent.
6.2 Analysis.
The following items
are needed for sample analysis:
6.2.1 Dilution System.
The dilution system
must be constructed such that all sample contacts are made of Teflon, glass, or
stainless steel. It must be capable of approximately a 9:1 dilution of the
sample.
6.2.2 Gas Chromatograph
(See Figure 15-2).
The gas chromatograph must have at least the following components:
6.2.2.1 Oven. Capable
of maintaining the separation column at the proper operating temperature ± 1
˚C.
6.2.2.2 Temperature
Gauge. To monitor column oven, detector, and exhaust temperature ± 1 ˚C.
6.2.2.3 Flow System.
Gas metering system to measure sample, fuel, combustion gas, and carrier gas
flows.
6.2.2.4 Flame
Photometric Detector.
6.2.2.4.1
Electrometer. Capable of full scale amplification of linear ranges of 10-9 to 10-4 amperes
full scale.
6.2.2.4.2 Power
Supply. Capable of delivering up to 750 volts.
6.2.2.5 Recorder.
Compatible with the output voltage range of the electrometer.
6.2.2.6 Rotary Gas
Valves. Multiport Teflon-lined valves equipped with sample loop. Sample loop volumes
shall be chosen to provide the needed analytical range. Teflon tubing and
fittings shall be used throughout to present an inert surface for sample gas.
The GC shall be calibrated with the sample loop used for sample analysis.
6.2.2.7 GC Columns. The
column system must be demonstrated to be capable of resolving three major
reduced sulfur compounds: H2S, COS, and CS2. To demonstrate that adequate resolution has been achieved, a
chromatogram of a calibration gas containing all three reduced sulfur compounds
in the concentration range of the applicable standard must be submitted.
Adequate resolution will be defined as base line separation of adjacent peaks
when the amplifier attenuation is set so that the smaller peak is at least 50
percent of full scale. Base line separation is defined as a return to zero (±5
percent) in the interval between peaks. Systems not meeting this criteria may
be considered alternate methods subject to the approval of the Administrator.
6.3 Calibration System
(See Figure 15-3).
The calibration system must contain the following components:
6.3.1 Flow System.
To measure air flow
over permeation tubes within 2 percent. Each flow meter shall be calibrated
after each complete test series with a wet-test meter. If the flow-measuring
device differs from the wet-test meter by more than 5 percent, the completed
test shall be discarded. Alternatively, use the flow data that will yield the
lowest flow measurement. Calibration with a wet-test meter before a test is
optional. Flow over the permeation device may also be determined using a soap
bubble flow meter.
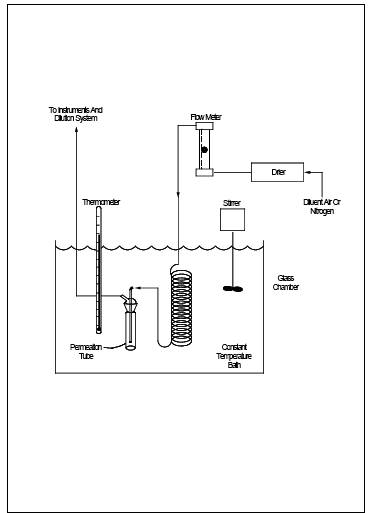
6.3.2 Constant Temperature Bath.
Device capable of
maintaining the permeation tubes at the calibration temperature within 0.1 ˚C.
6.3.3 Temperature Sensor.
Thermometer or
equivalent to monitor bath temperature within 0.1 ˚C.
7.0 Reagents and Standards.
7.1 Fuel. Hydrogen
gas (H2). Prepurified grade or better.
7.2 Combustion Gas.
Oxygen (O2) or air, research purity or better.
7.3 Carrier Gas.
Prepurified grade or better.
7.4 Diluent. Air
containing less than 0.5 ppmv total sulfur compounds and less than 10 ppmv each
of moisture and total hydrocarbons.
7.5 Calibration
Gases.
7.5.1 Permeation
Devices. One each of H2S, COS, and CS2,
gravimetrically calibrated and certified at some convenient operating
temperature. These tubes consist of hermetically sealed FEP Teflon tubing in
which a liquefied gaseous substance is enclosed. The enclosed gas permeates
through the tubing wall at a constant rate. When the temperature is constant,
calibration gases covering a wide range of known concentrations can be
generated by varying and accurately measuring the flow rate of diluent gas
passing over the tubes. These calibration gases are used to calibrate the
GC/FPD system and the dilution system.
7.5.2 Cylinder Gases.
Cylinder gases may be used as alternatives to permeation devices. The gases
must be traceable to a primary standard (such as permeation tubes) and not used
beyond the certification expiration date.
7.6 Citrate Buffer.
Dissolve 300 g of potassium citrate and 41 g of anhydrous citric acid in 1
liter of water. Alternatively, 284 g of sodium citrate may be substituted for
the potassium citrate. Adjust the pH to between 5.4 and 5.6 with potassium
citrate or citric acid, as required.
8.0 Sample Collection, Preservation, Transport, and Storage.
8.1 Pretest Procedures.
After the complete
measurement system has been set up at the site and deemed to be operational,
the following procedures should be completed before sampling is initiated. These
procedures are not required, but would be helpful in preventing any problem
which might occur later to invalidate the entire test.
8.1.1 Leak-Check.
Appropriate
leak-check procedures should be employed to verify the integrity of all
components, sample lines, and connections. The following procedure is
suggested: For components upstream of the sample pump, attach the probe end of
the sample line to a manometer or vacuum gauge, start the pump and pull a
vacuum greater than 50 mm (2 in.) Hg, close off the pump outlet, and then stop
the pump and ascertain that there is no leak for 1 minute. For components after
the pump, apply a slight positive pressure and check for leaks by applying a
liquid (detergent in water, for example) at each joint. Bubbling indicates the
presence of a leak. As an alternative to the initial leak-test, the sample line
loss test described in Section 8.3.1 may be performed to verify the integrity
of components.
8.1.2 System Performance.
Since the complete
system is calibrated at the beginning and end of each day of testing, the
precise calibration of each component is not critical. However, these
components should be verified to operate properly. This verification can be
performed by observing the response of flow meters or of the GC output to
changes in flow rates or calibration gas concentrations, respectively, and
ascertaining the response to be within predicted limits. If any component or
the complete system fails to respond in a normal and predictable manner, the
source of the discrepancy should be identified and corrected before proceeding.
8.2 Sample Collection and Analysis.
8.2.1 After
performing the calibration procedures outlined in Section 10.0, insert the
sampling probe into the test port ensuring that no dilution air enters the
stack through the port. Begin sampling and dilute the sample approximately 9:1
using the dilution system. Note that the precise dilution factor is the one
determined in Section 10.4. Condition the entire system with sample for a
minimum of 15 minutes before beginning the analysis. Inject aliquots of the
sample into the GC/FPD analyzer for analysis. Determine the concentration of
each reduced sulfur compound directly from the calibration curves or from the
equation for the least-squares line.
8.2.2 If reductions
in sample concentrations are observed during a sample run that cannot be
explained by process conditions, the sampling must be interrupted to determine
if the probe or filter is clogged with particulate matter. If either is found
to be clogged, the test must be stopped and the results up to that point
discarded. Testing may resume after cleaning or replacing the probe and filter.
After each run, the probe and filter shall be inspected and, if necessary,
replaced.
8.2.3 A sample run is
composed of 16 individual analyses (injects) performed over a period of not
less than 3 hours or more than 6 hours.
8.3 Post-Test Procedures.
8.3.1 Sample Line Loss.
A known concentration
of H2S at the level of the applicable standard, ±20
percent, must be introduced into the sampling system at the opening of the
probe in sufficient quantities to ensure that there is an excess of sample
which must be vented to the atmosphere. The sample must be transported through
the entire sampling system to the measurement system in the same manner as the
emission samples. The resulting measured concentration is compared to the known
value to determine the sampling system loss. For sampling losses greater than
20 percent, the previous sample run is not valid. Sampling losses of 0-20
percent must be corrected by dividing the resulting sample concentration by the
fraction of recovery. The known gas sample may be calibration gas as described
in Section 7.5. Alternatively, cylinder gas containing H2S mixed in nitrogen and verified according to Section 7.1.4 of
Method 16A may be used. The optional pretest procedures provide a good
guideline for determining if there are leaks in the sampling system.
8.3.2 Determination of Calibration Drift.
After each run, or
after a series of runs made within a 24-hour period, perform a partial
recalibration using the procedures in Section 10.0. Only H2S (or other permeant) need be used to recalibrate the GC/FPD
analysis system and the dilution system. Compare the calibration curves
obtained after the runs to the calibration curves obtained under Section 10.3.
The calibration drift should not exceed the limits set forth in Section 13.4.
If the drift exceeds this limit, the intervening run or runs should be
considered invalid. As an option, the calibration data set which gives the
highest sample values may be chosen by the tester.
9.0 Quality Control.
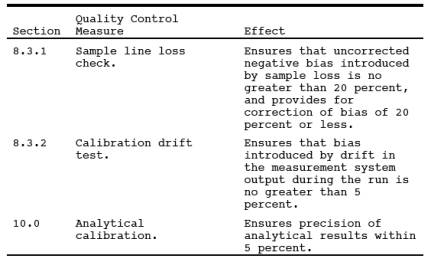
10.0 Calibration and Standardization.
Prior to any sampling
run, calibrate the system using the following procedures. (If more than one run
is performed during any 24-hour period, a calibration need not be performed
prior to the second and any subsequent runs. However, the calibration drift
must be determined as prescribed in Section 8.3.2 after the last run is made
within the 24-hour period.)
NOTE: This section outlines steps to be followed for
use of the GC/FPD and the dilution system. The calibration procedure does not
include detailed instructions because the operation of these systems is complex,
and it requires an understanding of the individual system being used. Each
system should include a written operating manual describing in detail the
operating procedures associated with each component in the measurement system.
In addition, the operator should be familiar with the operating principles of
the components, particularly the GC/FPD. The references in Section 16.0 are
recommended for review for this purpose.
10.1 Calibration Gas Permeation Tube Preparation.
10.1.1 Insert the
permeation tubes into the tube chamber. Check the bath temperature to assure
agreement with the calibration temperature of the tubes within 0.1 ˚C. Allow 24
hours for the tubes to equilibrate. Alternatively, equilibration may be
verified by injecting samples of calibration gas at 1-hour intervals. The
permeation tubes can be assumed to have reached equilibrium when consecutive
hourly samples agree within 5 percent of their mean.
10.1.2 Vary the
amount of air flowing over the tubes to produce the desired concentrations for
calibrating the analytical and dilution systems. The air flow across the tubes
must at all times exceed the flow requirement of the analytical systems. The
concentration in ppmv generated by a tube containing a specific permeant can be
calculated using Equation 15-1 in Section 12.2.
10.2 Calibration of Analytical System.
Generate a series of
three or more known concentrations spanning the linear range of the FPD
(approximately 0.5 to 10 ppmv for a 1-ml sample) for each of the three major
sulfur compounds. Bypassing the
dilution system, inject these standards into the GC/FPD and monitor the
responses until three consecutive injections for each concentration agree
within 5 percent of their mean. Failure to attain this precision indicates a
problem in the calibration or analytical system. Any such problem must be
identified and corrected before proceeding.
10.3 Calibration Curves.
Plot the GC/FPD
response in current (amperes) versus their causative concentrations in ppmv on
log-log coordinate graph paper for each sulfur compound. Alternatively, a
least-squares equation may be generated from the calibration data using
concentrations versus the appropriate instrument response units.
10.4 Calibration of Dilution System.
Generate a known
concentration of H2S using the permeation tube system. Adjust the
flow rate of diluent air for the first dilution stage so that the desired level
of dilution is approximated. Inject the diluted calibration gas into the GC/FPD
system until the results of three consecutive injections for each dilution
agree within 5 percent of their mean. Failure to attain this precision in this
step is an indication of a problem in the dilution system. Any such problem
must be identified and corrected before proceeding. Using the calibration data
for H2S (developed under Section 10.3), determine the
diluted calibration gas concentration in ppmv. Then calculate the dilution
factor as the ratio of the calibration gas concentration before dilution to the
diluted calibration gas concentration determined under this section. Repeat
this procedure for each stage of dilution required. Alternatively, the GC/FPD
system may be calibrated by generating a series of three or more concentrations
of each sulfur compound and diluting these samples before injecting them into
the GC/FPD system. These data will then serve as the calibration data for the
unknown samples and a separate determination of the dilution factor will not be
necessary. However, the precision requirements are still applicable.
11.0 Analytical Procedure.
Sample collection and
analysis are concurrent for this method (see Section 8.0).
12.0 Data Analysis and Calculations.
12.1 Nomenclature.
C = Concentration of
permeant produced, ppmv.
COS = Carbonyl
sulfide concentration, ppmv.
CS2 = Carbon disulfide concentration, ppmv.
d = Dilution factor,
dimensionless.
H2S = Hydrogen sulfide concentration, ppmv.
K = 24.04 L/g mole.
(Gas constant at 20˚C and 760 mm Hg)
L = Flow rate, L/min,
of air over permeant 20˚C, 760 mm Hg.
M = Molecular weight
of the permeant, g/g-mole.
N = Number of
analyses performed.
Pr = Permeation rate of the tube, µg/min.
12.2 Permeant
Concentration. Calculate the concentration generated by a tube containing a
specific permeant (see Section 10.1) using the following equation:
![]()
12.3 Calculation of
SO2 Equivalent. SO2 equivalent
will be determined for each analysis made by summing the concentrations of each
reduced sulfur compound resolved during the given analysis. The SO2 equivalent is expressed as SO2 in ppmv.
![]()
12.4 Average SO2 Equivalent. This is determined using the following equation.
Systems that do not remove moisture from the sample but condition the gas to
prevent condensation must correct the average SO2 equivalent
for the fraction of water vapor present. This is not done under applications
where the emission standard is not specified on a dry basis.
![]()
where:
Avg SO2 equivalent = Average SO2 equivalent
in ppmv, dry basis.
Average SO2 equivalent i
= SO2 in
ppmv as determined by Equation 15-2.
13.0 Method Performance.
13.1 Range.
Coupled with a GC
system using a 1-ml sample size, the maximum limit of the FPD for each sulfur
compound is approximately 10 ppmv. It may be necessary to dilute samples from
sulfur recovery plants a hundredfold (99:1), resulting in an upper limit of
about 1000 ppmv for each compound.
13.2 Sensitivity.
The minimum
detectable concentration of the FPD is also dependent on sample size and would
be about 0.5 ppmv for a 1-ml sample.
13.3 Calibration Precision.
A series of three
consecutive injections of the same calibration gas, at any dilution, shall
produce results which do not vary by more than 5 percent from the mean of the
three injections.
13.4 Calibration Drift.
The calibration drift
determined from the mean of three injections made at the beginning and end of
any run or series of runs within a 24-hour period shall not exceed 5 percent.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
1. O'Keeffe, A.E.,
and G.C. Ortman. "Primary Standards for Trace Gas Analysis." Anal.
Chem. 38,760. 1966.
2. Stevens, R.K.,
A.E. O'Keeffe, and G.C. Ortman. "Absolute Calibration of a Flame
Photometric Detector to Volatile Sulfur Compounds at Sub-Part-Per-Million
Levels." Environmental Science and Technology 3:7. July 1969.
3. Mulik, J.D., R.K.
Stevens, and R. Baumgardner. "An Analytical System Designed to Measure
Multiple Malodorous Compounds Related to Kraft Mill Activities." Presented
at the 12th Conference on Methods in Air Pollution and Industrial Hygiene
Studies, University of Southern California, Los Angeles, CA, April 6-8, 1971.
4. Devonald, R.H.,
R.S. Serenius, and A.D. McIntyre. "Evaluation of the Flame Photometric
Detector for Analysis of Sulfur Compounds." Pulp and Paper Magazine of
Canada, 73,3. March 1972.
5. Grimley, K.W.,
W.S. Smith, and R.M. Martin. "The Use of a Dynamic Dilution System in the
Conditioning of Stack Gases for Automated Analysis by a Mobile Sampling
Van." Presented at the 63rd Annual APCA Meeting in St. Louis, MO. June
14-19, 1970.
6. General Reference.
Standard Methods of Chemical Analysis Volume III-A and III-B: Instrumental
Analysis. Sixth Edition. Van Nostrand Reinhold Co.
17.0 Tables, Diagrams, Flowcharts, and Validation Data.
Figure 15-1. Sampling
and Dilution Apparatus.
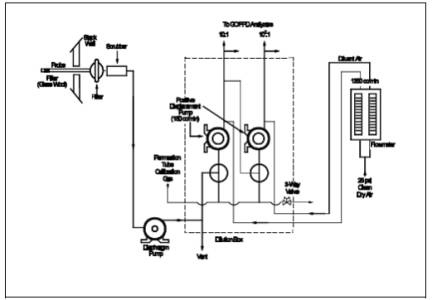
Figure 15-2. Gas
Chromatographic Flame Photometric Analyzer.
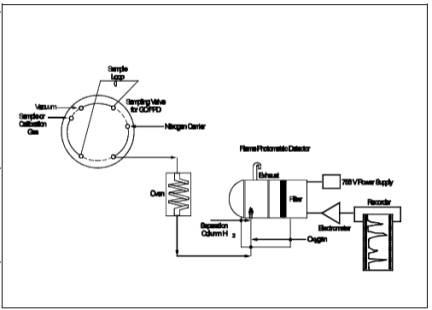
Figure 15-3.
Apparatus for Field Calibration.