METHOD 5F -
DETERMINATION OF NONSULFATE PARTICULATE MATTER EMISSIONS FROM STATIONARY
SOURCES
NOTE: This method does not include all of the
specifications (e.g.,
equipment and supplies) and procedures (e.g., sampling and analytical) essential to its
performance. Some material is incorporated by reference from other methods in
this part. Therefore, to obtain reliable results, persons using this method
should have a thorough knowledge of at least the following additional test
methods: Method 1, Method 2,
Method 3, and Method 5.
6.1 Sample Collection
and Recovery.
7.3.2 Stock Standard
Solution, 1 mg (NH4)2SO4/ml.
7.3.3 Working Standard
Solution
7.3.6 Phenolphthalein
Indicator.
8.0 Sample Collection,
Preservation, Storage, and Transport.
9.1 Miscellaneous
Quality Control Measures.
9.2 Volume Metering
System Checks.
10.0 Calibration and
Standardization.
10.1 Determination of
Ion Chromatograph Calibration Factor S.
12.0 Data Analysis and
Calculations.
12.2 Water Blank
Concentration.
12.3 Mass of Ammonium
Sulfate.
12.4 Mass of Nonsulfate
Particulate Matter.
13.0 Method Performance.
[Reserved]
14.0 Pollution
Prevention. [Reserved]
15.0 Waste Management.
[Reserved]
18.0 Tables, Diagrams,
Flowcharts, and Validation Data.
[Reserved]
1.0 Scope and Applications.
1.1 Analyte.
Nonsulfate particulate matter (PM). No CAS number assigned.
1.2 Applicability.
This method is applicable for the determination of nonsulfate PM emissions from
stationary sources. Use of this method must be specified by an applicable
subpart of the standards, or approved by the Administrator for a particular
application.
1.3 Data Quality
Objectives. Adherence to the requirements of this method will enhance the
quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method.
Particulate matter is
withdrawn isokinetically from the source and collected on a filter maintained
at a temperature in the range 160 ± 14 °C (320 ± 25 °F). The collected sample
is extracted with water. A portion of the extract is analyzed for sulfate
content by ion chromatography. The remainder is neutralized with ammonium
hydroxide (NH4OH), dried, and weighed. The weight of sulfate
in the sample is calculated as ammonium sulfate [(NH4)2SO4], and is
subtracted from the total particulate weight; the result is reported as
nonsulfate particulate matter.
3.0 Definitions. [Reserved]
4.0 Interferences. [Reserved]
5.0 Safety.
5.1 Disclaimer. This
method may involve hazardous materials, operations, and equipment. This test
method may not address all of the safety problems associated with its use. It
is the responsibility of the user of this test method to establish appropriate
safety and health practices and to determine the applicability of regulatory
limitations prior to performing this
test method.
6.0 Equipment and Supplies.
6.1 Sample Collection and Recovery.
Same as Method 5, Sections 6.1 and 6.2, respectively.
6.2 Sample Analysis.
Same as Method 5, Section 6.3, with the addition of the
following:
6.2.1 Erlenmeyer
Flasks. 125-ml, with ground glass joints.
6.2.2 Air Condenser.
With ground glass joint compatible with the Erlenmeyer flasks.
6.2.3 Beakers.
600-ml.
6.2.4 Volumetric
Flasks. 1-liter, 500-ml (one for each sample), 200-ml, and 50-ml (one for each
sample and standard).
6.2.5 Pipet. 5-ml
(one for each sample and standard).
6.2.6
Ion Chromatograph. The ion chromatograph should have at least the following
components.
6.2.6.1 Columns. An
anion separation column or other column capable of resolving the sulfate ion
from other species present and a standard anion suppressor column. Suppressor
columns are produced as proprietary items; however, one can be produced in the
laboratory using the resin available from BioRad Company, 32nd and Griffin
Streets, Richmond, California. Other systems which do not use suppressor
columns may also be used.
6.2.6.2 Pump. Capable
of maintaining a steady flow as required by the system.
6.2.6.3 Flow Gauges.
Capable of measuring the specified system flow rate.
6.2.6.4 Conductivity
Detector.
6.2.6.5 Recorder.
Compatible with the output voltage range of the detector.
7.0 Reagents and Standards.
Unless otherwise
indicated, it is intended that all reagents conform to the specifications
established by the Committee on Analytical Reagents of the American Chemical Society,
where such specifications are available; otherwise, use the best available
grade.
7.1 Sample Collection.
Same as Method 5, Section 7.1.
7.2 Sample Recovery.
Same as Method 5, Section 7.2, with the addition of the
following:
7.2.1
Water. Deionized distilled, to conform to ASTM D 1193-77 or 91 Type 3 incorporated by reference - see
§60.17). The potassium permanganate (KMnO4) test for
oxidizable organic matter may be omitted when high concentrations of organic
matter are not expected to be present.
7.3 Analysis.
Same as Method 5,
Section 7.3, with the addition of the following:
7.3.1 Water.
Same as in Section
7.2.1.
7.3.2 Stock Standard Solution, 1 mg (NH4)2SO4/ml.
Dry an adequate
amount of primary standard grade ammonium sulfate [(NH4)2SO4] at 105
to 110 °C (220 to 230 °F) for a minimum of 2 hours before preparing the
standard solution. Then dissolve exactly 1.000 g of dried (NH4)2SO4 in water
in a 1-liter volumetric flask, and dilute to 1 liter. Mix well.
7.3.3 Working Standard Solution
25 µg (NH4)2SO4/ml. Pipet
5 ml of the stock standard solution into a 200-ml volumetric flask. Dilute to
200 ml with water.
7.3.4 Eluent Solution.
Weigh 1.018 g of
sodium carbonate (Na2CO3) and
1.008 g of sodium bicarbonate (NaHCO3), and
dissolve in 4 liters of water. This solution is 0.0024 M Na2CO3/0.003 M NaHCO3.
Other eluents appropriate to the column type and capable of resolving sulfate
ion from other species present may be used.
7.3.5 Ammonium Hydroxide.
Concentrated, 14.8 M.
7.3.6 Phenolphthalein Indicator.
3,3-Bis(4-
hydroxyphenyl)-1-(3H)-isobenzo-furanone. Dissolve 0.05 g in 50 ml of ethanol
and 50 ml of water.
8.0 Sample Collection, Preservation, Storage, and Transport.
Same as Method 5, Section 8.0, with the exception of the
following:
8.1 Sampling Train Operation.
Same as Method 5, Section 8.5, except that the
probe outlet and filter temperatures shall be maintained at 160 ± 14 °C (320 ±
25 °F).
8.2 Sample Recovery.
Same as Method 5, Section 8.7, except that the
recovery solvent shall be water instead of acetone, and a clean filter from the
same lot as those used during testing shall be saved for analysis as a blank.
9.0 Quality Control.
9.1 Miscellaneous Quality Control Measures.
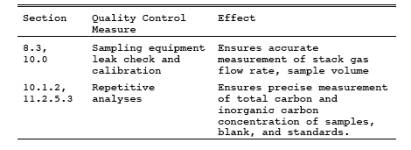
9.2 Volume Metering System Checks.
Same as Method 5, Section 9.2.
10.0 Calibration and Standardization.
Same as Method 5, Section 10.0, with the addition of
the following:
10.1 Determination of Ion Chromatograph Calibration Factor S.
Prepare a series of
five standards by adding 1.0, 2.0, 4.0, 6.0, and 10.0 ml of working standard
solution (25 µg/ml) to a series of five 50-ml volumetric flasks. (The standard
masses will equal 25, 50, 100, 150, and 250 µg.) Dilute each flask to the mark with water, and mix well.
Analyze each standard according to the chromatograph manufacturer's
instructions. Take peak height measurements with symmetrical peaks; in all
other cases, calculate peak areas. Prepare or calculate a linear regression
plot of the standard masses in µg (x-axis) versus their responses (y-axis).
From this line, or equation, determine the slope and calculate its reciprocal
which is the calibration factor, S. If any point deviates from the line by more
than 7 percent of the concentration at that point, remake and reanalyze that
standard. This deviation can be determined by multiplying S times the response
for each standard. The resultant concentrations must not differ by more than 7
percent from each known standard mass (i.e., 25, 50, 100, 150, and 250 µg).
10.2 Conductivity Detector.
Calibrate according
to manufacturer's specifications prior to initial use.
11.0 Analytical Procedure.
11.1 Sample Extraction.
11.1.1 Note on the
analytical data sheet, the level of the liquid in the container, and whether
any sample was lost during shipment. If a noticeable amount of leakage has
occurred, either void the sample or use methods, subject to the approval of the
Administrator, to correct the final results.
11.1.2 Cut the filter
into small pieces, and place it in a 125-ml Erlenmeyer flask with a ground
glass joint equipped with an air condenser. Rinse the shipping container with
water, and pour the rinse into the flask. Add additional water to the flask
until it contains about 75 ml, and place the flask on a hot plate. Gently
reflux the contents for 6 to 8 hours. Cool the solution, and transfer it to a
500-ml volumetric flask. Rinse the Erlenmeyer flask with water, and transfer
the rinsings to the volumetric flask including the pieces of filter.
11.1.3
Transfer the probe rinse to the same 500-ml volumetric flask with the filter
sample. Rinse the sample bottle with water, and add the rinsings to the
volumetric flask. Dilute the contents of the flask to the mark with water.
11.1.4 Allow the contents
of the flask to settle until all solid material is at the bottom of the flask.
If necessary, remove and centrifuge a portion of the sample.
11.1.5 Repeat the
procedures outlined in Sections 11.1.1 through 11.1.4 for each sample and for
the filter blank.
11.2 Sulfate (SO4) Analysis.
11.2.1 Prepare a
standard calibration curve according to the procedures outlined in Section
10.1.
11.2.2 Pipet 5 ml of
the sample into a 50-ml volumetric flask, and dilute to 50 ml with water.
(Alternatively, eluent solution may be used instead of water in all sample,
standard, and blank dilutions.) Analyze the set of standards followed by the
set of samples, including the filter blank, using the same injection volume
used for the standards.
11.2.3 Repeat the
analyses of the standards and the samples, with the standard set being done
last. The two peak height or peak area responses for each sample must agree
within 5 percent of their arithmetic mean for the analysis to be valid. Perform
this analysis sequence on the same day. Dilute any sample and the blank with
equal volumes of water if the concentration exceeds that of the highest
standard.
11.2.4 Document each
sample chromatogram by listing the following analytical parameters: injection
point, injection volume, sulfate retention time, flow rate, detector
sensitivity setting, and recorder chart speed.
11.3 Sample Residue.
11.3.1 Transfer the
remaining contents of the volumetric flask to a tared 600-ml beaker or similar
container. Rinse the volumetric flask with water, and add the rinsings to the
tared beaker. Make certain that all particulate matter is transferred to the
beaker. Evaporate the water in an oven at 105 °C (220 °F) until only about 100
ml of water remains. Remove the beakers from the oven, and allow them to cool.
11.3.2 After the
beakers have cooled, add five drops of phenolphthalein indicator, and then add
concentrated ammonium hydroxide until the solution turns pink. Return the
samples to the oven at 105 °C (220 °F), and evaporate the samples to dryness.
Cool the samples in a desiccator, and weigh the samples to constant weight.
12.0 Data Analysis and Calculations.
Same as Method 5, Section 12.0, with the addition of the
following:
12.1 Nomenclature.
Cw = Water blank residue concentration, mg/ml.
F = Dilution factor
(required only if sample dilution was needed to reduce the concentration into
the range of calibration).
Hs = Arithmetic mean response of duplicate sample analyses, mm for
height or mm2 for area.
Hb = Arithmetic mean response of duplicate filter blank analyses, mm
for height or mm2 for area.
mb = Mass of beaker used to dry sample, mg.
mf = Mass of sample filter, mg.
mn = Mass of nonsulfate particulate matter in the sample as collected,
mg.
ms = Mass of ammonium sulfate in the sample as collected, mg.
mt = Mass of beaker, filter, and dried sample, mg.
mw = Mass of residue after evaporation of water blank, mg.
S = Calibration
factor, µg/mm.
Vb = Volume of water blank, ml.
Vs = Volume of sample collected, 500 ml.
12.2 Water Blank Concentration.
![]()
12.3 Mass of Ammonium Sulfate.
![]()
where:
100 = Aliquot factor,
495 ml/5 ml
1000 = Constant,
µg/mg
12.4 Mass of Nonsulfate Particulate Matter.
![]()
13.0 Method Performance. [Reserved]
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 Alternative Procedures
16.1 The following
procedure may be used as an alternative to the procedure in Section
11.0
16.1.1 Apparatus.
Same as for Method 6, Sections 6.3.3 to
6.3.6 with the following additions.
16.1.1.1 Beakers.
250-ml, one for each sample, and 600-ml.
16.1.1.2 Oven.
Capable of maintaining temperatures of 75 ± 5°C (167 ± 9°F) and 105 ± 5°C (221
± 9°F).
16.1.1.3 Buchner
Funnel.
16.1.1.4 Glass
Columns. 25-mm x 305-mm (1-in. x 12-in.) with Teflon stopcock.
16.1.1.5 Volumetric
Flasks. 50-ml and 500-ml, one set for each sample, and 100-ml, 200-ml, and
1000-ml.
16.1.1.6 Pipettes.
Two 20-ml and one 200-ml, one set for each sample, and 5-ml.
16.1.1.7 Filter
Flasks. 500-ml.
16.1.1.8 Polyethylene
Bottle. 500-ml, one for each sample.
16.1.2 Reagents. Same
as Method 6, Sections 7.3.2 to 7.3.5 with
the following additions:
16.1.2.1 Water,
Ammonium Hydroxide, and Phenolphthalein. Same as Sections
7.2.1, 7.3.5, and 7.3.6 of this method, respectively.
16.1.2.2 Filter.
Glass fiber to fit Buchner funnel.
16.1.2.3 Hydrochloric
Acid (HCl), 1 m. Add 8.3 ml of concentrated HCl (12 M) to 50 ml of water in a
100-ml volumetric flask. Dilute to 100 ml with water.
16.1.2.4 Glass Wool.
16.1.2.5 Ion Exchange
Resin. Strong cation exchange resin, hydrogen form, analytical grade.
16.1.2.6 pH Paper.
Range of 1 to 7.
16.1.3 Analysis.
16.1.3.1 Ion Exchange
Column Preparation. Slurry the resin with 1 M HCl in a 250-ml beaker, and allow
to stand overnight. Place 2.5 cm (1 in.) of glass wool in the bottom of the
glass column. Rinse the slurried resin twice with water. Resuspend the resin in
water, and pour sufficient resin into the column to make a bed 5.1 cm (2 in.)
deep. Do not allow air bubbles to become entrapped in the resin or glass wool to
avoid channeling, which may produce erratic results. If necessary, stir the
resin with a glass rod to remove air bubbles, after the column has been
prepared, never let the liquid level fall below the top of the upper glass wool
plug. Place a 2.5-cm (1-in.) plug of glass wool on top of the resin. Rinse the
column with water until the eluate gives a pH of 5 or greater as measured with
pH paper.
16.1.3.2 Sample
Extraction. Followup the procedure given in Section
11.1.3 except do not dilute the sample to 500 ml.
16.1.3.3 Sample
Residue.
16.1.3.3.1 Place at
least one clean glass filter for each sample in a Buchner funnel, and rinse the
filters with water. Remove the filters from the funnel, and dry them in an oven
at 105 ± 5°C (221 ± 9°F); then cool in a desiccator. Weigh each filter to
constant weight according to the procedure in Method
5, Section 11.0. Record the weight of each filter to the nearest 0.1 mg.
16.1.3.3.2 Assemble the
vacuum filter apparatus, and place one of the clean, tared glass fiber filters
in the Buchner funnel. Decant the liquid portion of the extracted sample
(Section 16.1.3.2) through the tared glass fiber filter into a clean, dry,
500-ml filter flask. Rinse all the particulate matter remaining in the
volumetric flask onto the glass fiber filter with water. Rinse the particulate
matter with additional water. Transfer the filtrate to a 500-ml volumetric
flask, and dilute to 500 ml with water. Dry the filter overnight at 105 ± 5°C
(221 ± 9°F), cool in a desiccator, and weigh to the nearest 0.1 mg.
16.1.3.3.3 Dry a
250-ml beaker at 75 ± 5°C (167 ± 9°F), and cool in a desiccator; then weigh to
constant weight to the nearest 0.1 mg. Pipette 200 ml of the filtrate that was
saved into a tared 250-ml beaker; add five drops of phenolphthalein indicator
and sufficient concentrated ammonium hydroxide to turn the solution pink.
Carefully evaporate the contents of the beaker to dryness at 75 ± 5°C (167 ±
9°F). Check for dryness every 30 minutes. Do not continue to bake the sample
once it has dried. Cool the sample in a desiccator, and weigh to constant
weight to the nearest 0.1 mg.
16.1.3.4 Sulfate
Analysis. Adjust the flow rate through the ion exchange column to 3 ml/min.
Pipette a 20-ml aliquot of the filtrate onto the top of the ion exchange
column, and collect the eluate in a 50-ml volumetric flask. Rinse the column
with two 15-ml portions of water. Stop collection of the eluate when the volume
in the flask reaches 50-ml. Pipette a 20-ml aliquot of the eluate into a 250-ml
Erlenmeyer flask, add 80 ml of 100 percent isopropanol and two to four drops of
thorin indicator, and titrate to a pink end point using 0.0100 N barium
perchlorate. Repeat and average the titration volumes. Run a blank with each
series of samples. Replicate titrations must agree within 1 percent or 0.2 ml,
whichever is larger. Perform the ion exchange and titration procedures on
duplicate portions of the filtrate. Results should agree within 5 percent.
Regenerate or replace the ion exchange resin after 20 sample aliquots have been
analyzed or if the end point of the titration becomes unclear.
NOTE: Protect the 0.0100 N barium perchlorate solution
from evaporation at all times.
16.1.3.5 Blank
Determination. Begin with a sample of water of the same volume as the samples
being processed and carry it through the analysis steps described in Sections
16.1.3.3 and 16.1.3.4. A blank value larger than 5 mg should not be subtracted
from the final particulate matter mass. Causes for large blank values should be
investigated and any problems resolved before proceeding with further analyses.
16.1.4 Calibration.
Calibrate the barium perchlorate solutions as in Method
6, Section 10.5.
16.1.5 Calculations.
16.1.5.1
Nomenclature. Same as Section 12.1 with the
following additions:
ma = Mass of clean analytical filter, mg.
md = Mass of dissolved particulate matter, mg.
me = Mass of beaker and dissolved particulate matter after evaporation
of filtrate, mg.
mp = Mass of insoluble particulate matter, mg.
mr = Mass of analytical filter, sample filter, and insoluble
particulate matter, mg.
mbk = Mass of nonsulfate particulate matter in blank
sample, mg.
mn = Mass of nonsulfate particulate matter, mg.
ms = Mass of Ammonium sulfate, mg.
N = Normality of
Ba(Cl04) titrant, meq/ml.
Va = Volume of aliquot taken for titration, 20 ml.
Vc = Volume of titrant used for titration blank, ml.
Vd = Volume of filtrate evaporated, 200 ml.
Ve = Volume of eluate collected, 50 ml.
Vf = Volume of extracted sample, 500 ml.
Vi = Volume of filtrate added to ion exchange column, 20 ml.
Vt = Volume of Ba(C104)2 titrant, ml.
W = Equivalent weight
of ammonium sulfate, 66.07 mg/meq.
16.1.5.2 Mass of
Insoluble Particulate Matter.
mp = mr-ma-mf Eq. 5F-4
16.1.5.3 Mass of
Dissolved Particulate Matter.
md = (me - (Vf/Vd)mb) Eq.
5F-5
16.1.5.4 Mass of
Ammonium Sulfate.
16.1.5.5 Mass of Nonsulfate
Particulate Matter.
mn = mp + md - ms - mbk Eq. 5F-7
17.0 References.
Same as Method 5, Section 17.0, with the addition of the
following:
1. Mulik, J.D. and E.
Sawicki. Ion Chromatographic Analysis of Environmental Pollutants. Ann Arbor,
Ann Arbor Science Publishers, Inc. Vol. 2, 1979.
2. Sawicki, E., J.D.
Mulik, and E. Wittgenstein. Ion Chromatographic Analysis of Environmental Pollutants.
Ann Arbor, Ann Arbor Science Publishers, Inc. Vol. 1. 1978.
3. Siemer, D.D.
Separation of Chloride and Bromide from Complex Matrices Prior to Ion
Chromatographic Determination. Analytical Chemistry 52(12): 1874-1877. October
1980.
4. Small, H., T.S.
Stevens, and W.C. Bauman. Novel Ion Exchange Chromatographic Method Using
Conductimetric Determination. Analytical Chemistry. 47(11):1801. 1975.
18.0 Tables, Diagrams, Flowcharts, and Validation Data. [Reserved]