METHOD 25 -
DETERMINATION OF TOTAL GASEOUS NONMETHANE
ORGANIC EMISSIONS
AS CARBON
4.1 Carbon Dioxide and
Water Vapor.
7.5 Quality Assurance
Audit Samples.
8.0 Sample Collection,
Preservation, Transport, and Storage.
8.1 Sampling Equipment
Preparation.
8.1.1 Condensate Trap
Cleaning.
8.1.2 Sample Tank
Evacuation and Leak-Check.
8.1.3 Sampling Train
Assembly.
8.4 Sample Storage and
Transport.
10.0 Calibration and
Standardization.
10.1 Initial
Performance Checks.
10.1.1 Condensate
Recovery Apparatus.
10.2 NMO Analyzer Daily
Calibration.
10.3 Sample Tank and
ICV Volume.
12.0 Data Analysis and
Calculations.
14.0 Pollution
Prevention. [Reserved]
15.0 Waste Management.
[Reserved]
17.0 Tables, Diagrams,
Flowcharts, and Validation Data.
1.0 Scope and Application.
1.1 Analytes.

1.2 Applicability.
1.2.1 This method is
applicable for the determination of volatile organic compounds (VOC) (measured
as total gaseous non-methane organics (TGNMO) and reported as carbon) in stationary
source emissions. This method is not applicable for the determination of
organic particulate matter.
1.2.2 This method is
not the only method that applies to the measurement of VOC. Costs, logistics,
and other practicalities of source testing may make other test methods more
desirable for measuring VOC contents of certain effluent streams. Proper
judgment is required in determining the most applicable VOC test method. For
example, depending upon the molecular composition of the organics in the effluent
stream, a totally automated semi-continuous non-methane organics (NMO) analyzer
interfaced directly to the source may yield accurate results. This approach has
the advantage of providing emission data semi-continuously over an extended
time period.
1.2.3 Direct
measurement of an effluent with a flame ionization detector (FID) analyzer may
be appropriate with prior characterization of the gas stream and knowledge that
the detector responds predictably to the organic compounds in the stream. If
present, methane (CH4) will, of course, also be measured. The FID can
be used under any of the following limited conditions: (1) where only one
compound is known to exist; (2) when the organic compounds consist of only
hydrogen and carbon; (3) where the relative percentages of the compounds are
known or can be determined, and the FID responses to the compounds are known;
(4) where a consistent mixture of the compounds exists before and after
emission control and only the relative concentrations are to be assessed; or
(5) where the FID can be calibrated against mass standards of the compounds
emitted (solvent emissions, for example).
1.2.4 Another example
of the use of a direct FID is as a screening method. If there is enough
information available to provide a rough estimate of the analyzer accuracy, the
FID analyzer can be used to determine the VOC content of an uncharacterized gas
stream. With a sufficient buffer to account for possible inaccuracies, the
direct FID can be a useful tool to obtain the desired results without costly
exact determination.
1.2.5 In situations
where a qualitative/quantitative analysis of an effluent stream is desired or
required, a gas chromatographic FID system may apply. However, for sources
emitting numerous organics, the time and expense of this approach will be
formidable.
2.0 Summary of Method.
2.1 An emission
sample is withdrawn from the stack at a constant rate through a heated filter
and a chilled condensate trap by means of an evacuated sample tank. After
sampling is completed, the TGNMO are determined by independently analyzing the
condensate trap and sample tank fractions and combining the analytical results.
The organic content of the condensate trap fraction is determined by oxidizing
the NMO to carbon dioxide (CO2) and quantitatively
collecting in the effluent in an evacuated vessel; then a portion of the CO2 is reduced to CH4 and measured by an
FID. The organic content of the sample tank fraction is measured by injecting a
portion of the sample into a gas chromatographic column to separate the NMO
from carbon monoxide (CO), CO2, and CH4; the NMO are oxidized to CO2, reduced
to CH4, and measured by an FID. In this manner, the
variable response of the FID associated with different types of organics is
eliminated.
3.0 Definitions. [Reserved]
4.0 Interferences.
4.1 Carbon Dioxide and Water Vapor.
When carbon dioxide
(CO2) and water vapor are present together in the
stack, they can produce a positive bias in the sample. The magnitude of the
bias depends on the concentrations of CO2 and water
vapor. As a guideline, multiply the CO2 concentration,
expressed as volume percent, times the water vapor concentration. If this
product does not exceed 100, the bias can be considered insignificant. For
example, the bias is not significant for a source having 10 percent CO2 and 10 percent water vapor, but it might be significant for a
source having 10 percent CO2 and 20 percent water
vapor.
4.2. Particulate Matter.
Collection of organic
particulate matter in the condensate trap would produce a positive bias. A
filter is included in the sampling equipment to minimize this bias.
5.0 Safety.
5.1 Disclaimer. This
method may involve hazardous materials, operations, and equipment. This test
method may not address all of the safety problems associated with its use. It
is the responsibility of the user of this test method to establish appropriate
safety and health practices and determine the applicability of regulatory
limitations prior to performing this test method.
6.0 Equipment and Supplies.
6.1 Sample Collection.
The sampling system
consists of a heated probe, heated filter, condensate trap, flow control
system, and sample tank (see Figure 25-1). The TGNMO sampling equipment can be
constructed from commercially available components and components fabricated in
a machine shop. The following equipment is required:
6.1.1 Heated Probe.
6.4-mm (1/4-in.) OD stainless steel tubing with a heating system capable of
maintaining a gas temperature at the exit end of at least 129˚C (265˚F). The
probe shall be equipped with a temperature sensor at the exit end to monitor
the gas temperature. A suitable probe is shown in Figure 25-1. The nozzle is an
elbow fitting attached to the front end of the probe while the temperature
sensor is inserted in the side arm of a tee fitting attached to the rear of the
probe. The probe is wrapped with a suitable length of high temperature heating
tape, and then covered with two layers of glass cloth insulation and one layer
of aluminum foil or an equivalent wrapping.
NOTE: If it is not possible to use a heating system
for safety reasons, an unheated system with an in-stack filter is a suitable
alternative.
6.1.2 Filter Holder.
25-mm (15/16-in.) ID Gelman filter holder with 303 stainless steel body and 316
stainless steel support screen with the Viton O-ring replaced by a Teflon
O-ring.
6.1.3 Filter Heating
System.
6.1.3.1 A metal box
consisting of an inner and an outer shell separated by insulating material with
a heating element in the inner shell capable of maintaining a gas temperature
at the filter of 121 ± 3 ˚C (250 ± 5 ˚F). The heating box shall include
temperature sensors to monitor the gas temperature immediately upstream and
immediately downstream of the filter.
6.1.3.2 A suitable
heating box is shown in Figure 25-2. The outer shell is a metal box that
measures 102 mm x 280 mm x 292 mm (4 in. x 11 in. x 11 1/2 in.), while the
inner shell is a metal box measuring 76 mm x 229 mm x 241 mm (3 in. x 9 in. x 9
1/2 in.). The inner box is supported by 13-mm (1/2-in.) phenolic rods. The void
space between the boxes is filled with ceramic fiber insulation which is sealed
in place by means of a silicon rubber bead around the upper sides of the box. A
removable lid made in a similar manner, with a 25-mm (1-in.) gap between the
parts is used to cover the heating chamber. The inner box is heated with a
250-watt cartridge heater, shielded by a stainless steel shroud. The heater is
regulated by a thermostatic temperature controller, which is set to maintain a
gas temperature of 121 ˚C (250 ˚F) as measured by the temperature sensor
upstream of the filter.
NOTE: If it is not possible to use a heating system
for safety reasons, an unheated system with an in-stack filter is a suitable
alternative.
6.1.4 Condensate Trap.
9.5-mm (d-in.) OD 316 stainless steel tubing bent into a U-shape. Exact
dimensions are shown in Figure 25-3. The tubing shall be packed with coarse
quartz wool, to a density of approximately 0.11 g/cm3 before bending. While the condensate trap is packed with dry ice in
the Dewar, an ice bridge may form between the arms of the condensate trap
making it difficult to remove the condensate trap. This problem can be
prevented by attaching a steel plate between the arms of the condensate trap in
the same plane as the arms to completely fill the intervening space.
6.1.5 Valve.
Stainless steel control valve for starting and stopping sample flow.
6.1.6 Metering Valve.
Stainless steel valve for regulating the sample flow rate through the sample
train.
6.1.7 Rate Meter.
Rotameter, or equivalent, capable of measuring sample flow in the range of 60
to 100 cm3/min (0.13 to 0.21 ft3/hr).
6.1.8 Sample Tank.
Stainless steel or aluminum tank with a minimum volume of 4 liters (0.14 ft3).
NOTE: Sample volumes greater than 4 liters may be
required for sources with low organic concentrations.
6.1.9 Mercury
Manometer. U-tube manometer or absolute pressure gauge capable of measuring
pressure to within 1 mm Hg in the range of 0 to 900 mm.
6.1.10 Vacuum Pump. Capable
of evacuating to an absolute pressure of 10 mm Hg.
6.2 Condensate Recovery.
The system for the
recovery of the organics captured in the condensate trap consists of a heat
source, an oxidation catalyst, a non-dispersive infrared (NDIR) analyzer, and
an intermediate collection vessel (ICV). Figure 25-4 is a schematic of a
typical system. The system shall be capable of proper oxidation and recovery,
as specified in Section 10.1.1. The following major components are required:
6.2.1 Heat Source.
Sufficient to heat the condensate trap (including probe) to a temperature of
200 ˚C (390 ˚F). A system using both a heat gun and an electric tube furnace is
recommended.
6.2.2 Heat Tape.
Sufficient to heat the connecting tubing between the water trap and the oxidation
catalyst to 100 ˚C (212 ˚F).
6.2.3 Oxidation
Catalyst. A suitable length of 9.5 mm (d-in.) OD Inconel 600 tubing packed with
15 cm (6 in.) of 3.2 mm (c-in.) diameter 19 percent chromia on alumina pellets.
The catalyst material is packed in the center of the catalyst tube with quartz
wool packed on either end to hold it in place.
6.2.4 Water Trap.
Leak-proof, capable of removing moisture from the gas stream.
6.2.5 Syringe Port. A
6.4-mm (1/4-in.) OD stainless steel tee fitting with a rubber septum placed in
the side arm.
6.2.6 NDIR Detector.
Capable of indicating CO2
concentration in the range of zero
to 5 percent, to monitor the progress of combustion of the organic compounds
from the condensate trap.
6.2.7 Flow-Control
Valve. Stainless steel, to maintain the trap conditioning system near
atmospheric pressure.
6.2.8 Intermediate
Collection Vessel. Stainless steel or aluminum, equipped with a female quick
connect. Tanks with nominal volumes of at least 6 liters (0.2 ft3) are recommended.
6.2.9 Mercury
Manometer. Same as described in Section 6.1.9.
6.2.10 Syringe. 10-ml
gas-tight glass syringe equipped with an appropriate needle.
6.2.11 Syringes.
10-µl and 50-µl liquid injection syringes.
6.2.12 Liquid Sample Injection
Unit. 316 Stainless steel U-tube fitted with an injection septum (see Figure
25-7).
6.3 Analysis.
6.3.1 NMO Analyzer.
The NMO analyzer is a
gas chromatograph (GC) with back-flush capability for NMO analysis and is
equipped with an oxidation catalyst, reduction catalyst, and FID. Figures 25-5 and 25-6 are schematics of a typical NMO analyzer. This semi-continuous GC/FID
analyzer shall be capable of: (1) separating CO, CO2, and CH4
from NMO, (2) reducing the CO2 to CH4 and quantifying as CH4, and (3) oxidizing the NMO to CO2,
reducing the CO2 to CH4 and
quantifying as CH4, according to Section 10.1.2. The analyzer
consists of the following major components:
6.3.1.1 Oxidation
Catalyst. A suitable length of 9.5-mm (d-in.) OD Inconel 600 tubing packed with
5.1 cm (2 in.) of 19 percent chromia on 3.2-mm (c-in.) alumina pellets. The
catalyst material is packed in the center of the tube supported on either side
by quartz wool. The catalyst tube must be mounted vertically in a 650 ˚C (1200
˚F) furnace. Longer catalysts mounted horizontally may be used, provided they
can meet the specifications of Section 10.1.2.1.
6.3.1.2 Reduction
Catalyst. A 7.6-cm (3-in.) length of 6.4-mm (1/4-in.) OD Inconel tubing fully
packed with 100- mesh pure nickel powder. The catalyst tube must be mounted
vertically in a 400 ˚C (750 ˚F) furnace.
6.3.1.3 Separation
Column(s). A 30-cm (1-ft) length of 3.2-mm (c-in.) OD stainless steel tubing
packed with 60/80 mesh Unibeads 1S followed by a 61-cm (2-ft) length of 3.2-mm
(c-in.) OD stainless steel tubing packed with 60/80 mesh Carbosieve G. The
Carbosieve and Unibeads columns must be baked separately at 200 ˚C (390 ˚F)
with carrier gas flowing through them for 24 hours before initial use.
6.3.1.4 Sample
Injection System. A single 10-port GC sample injection valve or a group of
valves with sufficient ports fitted with a sample loop properly sized to
interface with the NMO analyzer (1-cc loop recommended).
6.3.1.5 FID. An FID
meeting the following specifications is required:
6.3.1.5.1 Linearity.
A linear response (±5 percent) over the operating range as demonstrated by the
procedures established in Section 10.1.2.3.
6.3.1.5.2 Range. A
full scale range of 10 to 50,000 ppm CH4. Signal
attenuators shall be available to produce a minimum signal response of 10
percent of full scale.0
6.3.1.6 Data
Recording System. Analog strip chart recorder or digital integration system
compatible with the FID for permanently recording the analytical results.
6.3.2 Barometer.
Mercury, aneroid, or
other barometer capable of measuring atmospheric pressure to within 1 mm Hg.
6.3.3 Temperature Sensor.
Capable of measuring
the laboratory temperature within 1 ˚C (2 ˚F).
6.3.4 Vacuum Pump.
Capable of evacuating
to an absolute pressure of 10 mm Hg.
7.0 Reagents and Standards.
7.1 Sample Collection.
The following
reagents are required for sample collection:
7.1.1 Dry Ice. Solid
CO2, crushed.
7.1.2 Coarse Quartz
Wool. 8 to 15 µm.
7.1.3 Filters. Glass
fiber filters, without organic binder.
7.2 NMO Analysis.
The following gases
are required for NMO analysis:
7.2.1 Carrier Gases.
helium (He) and oxygen (O2) containing less than 1 ppm CO2 and less than 0.1 ppm hydrocarbon.
7.2.2 Fuel Gas.
Hydrogen (H2), at least 99.999 percent pure.
7.2.3 Combustion Gas.
Either air (less than 0.1 ppm total hydrocarbon content) or O2 (purity 99.99 percent or greater), as required by the detector.
7.3 Condensate Analysis.
The following are
required for condensate analysis:
7.3.1 Gases.
Containing less than 1 ppm carbon.
7.3.1.1 Air.
7.3.1.2 Oxygen.
7.3.2 Liquids. To
conform to the specifications established by the Committee on Analytical
Reagents of the American Chemical Society.
7.3.2.1 Hexane.
7.3.2.2 Decane.
7.4 Calibration.
For all calibration
gases, the manufacturer must recommend a maximum shelf life for each cylinder (i.e., the length of time the gas concentration is
not expected to change more than ±5 percent from its certified value). The date
of gas cylinder preparation, certified organic concentration, and recommended
maximum shelf life must be affixed to each cylinder before shipment from the
gas manufacturer to the buyer. The following calibration gases are required:
7.4.1 Oxidation
Catalyst Efficiency Check Calibration Gas. Gas mixture standard with nominal
concentration of 1 percent methane in air.
7.4.2 FID Linearity
and NMO Calibration Gases. Three gas mixture standards with nominal propane
concentrations of 20 ppm, 200 ppm, and 3000 ppm, in air.
7.4.3 CO2 Calibration Gases. Three gas mixture standards with nominal CO2 concentrations of 50 ppm, 500 ppm, and 1 percent, in air.
NOTE: Total NMO less than 1 ppm required for 1 percent
mixture.
7.4.4 NMO Analyzer
System Check Calibration Gases. Four calibration gases are needed as follows:
7.4.4.1 Propane
Mixture. Gas mixture standard containing (nominal) 50 ppm CO, 50 ppm CH4, 1 percent CO2, and 20 ppm C3H8, prepared in air.
7.4.4.2 Hexane. Gas
mixture standard containing (nominal) 50 ppm hexane in air.
7.4.4.3 Toluene. Gas
mixture standard containing (nominal) 20 ppm toluene in air.
7.4.4.4 Methanol. Gas
mixture standard containing (nominal) 100 ppm methanol in air.
7.5 Quality Assurance Audit Samples.
7.5.1 It is
recommended, but not required, that a performance audit sample be analyzed in
conjunction with the field samples. The audit sample should be in a suitable
sample matrix at a concentration similar to the actual field samples.
7.5.2 When making compliance
determinations, and upon availability, audit samples may be obtained from the
appropriate EPA Regional Office or from the responsible enforcement authority
and analyzed in conjunction with the field samples.
NOTE: The responsible enforcement authority should
be notified at least 30 days prior to the test date to allow sufficient time
for sample delivery.
8.0 Sample Collection, Preservation, Transport, and Storage.
8.1 Sampling Equipment Preparation.
8.1.1 Condensate Trap Cleaning.
Before its initial
use and after each use, a condensate trap should be thoroughly cleaned and
checked to ensure that it is not contaminated. Both cleaning and checking can
be accomplished by installing the trap in the condensate recovery system and
treating it as if it were a sample. The trap should be heated as described in
Section 11.1.3. A trap may be considered clean when the CO2 concentration in its effluent gas drops below 10 ppm. This check is
optional for traps that most recently have been used to collect samples which
were then recovered according to the procedure in Section 11.1.3.
8.1.2 Sample Tank Evacuation and Leak-Check.
Evacuate the sample
tank to 10 mm Hg absolute pressure or less. Then close the sample tank valve,
and allow the tank to sit for 60 minutes. The tank is acceptable if a change in
tank vacuum of less than 1 mm Hg is noted. The evacuation and leak-check may be
conducted either in the laboratory or the field.
8.1.3 Sampling Train Assembly.
Just before assembly,
measure the tank vacuum using a mercury manometer. Record this vacuum, the
ambient temperature, and the barometric pressure at this time. Close the sample
tank valve and assemble the sampling system as shown in Figure 25-1. Immerse
the condensate trap body in dry ice at least 30 minutes before commencing
sampling to improve collection efficiency. The point where the inlet tube joins
the trap body should be 2.5 to 5 cm (1 to 2 in.) above the top of the dry ice.
8.1.4 Pretest Leak-Check.
A pretest leak-check
is required. Calculate or measure the approximate volume of the sampling train
from the probe tip to the sample tank valve. After assembling the sampling
train, plug the probe tip, and make certain that the sample tank valve is
closed. Turn on the vacuum pump, and evacuate the sampling system from the
probe tip to the sample tank valve to an absolute pressure of 10 mm Hg or less.
Close the purge valve, turn off the pump, wait a minimum period of 10 minutes,
and recheck the indicated vacuum. Calculate the maximum allowable pressure
change based on a leak rate of 1 percent of the sampling rate using Equation
25-1, Section 12.2. If the measured pressure change exceeds the allowable,
correct the problem and repeat the leak-check before beginning sampling.
8.2 Sample Collection.
8.2.1 Unplug the
probe tip, and place the probe into the stack such that the probe is
perpendicular to the duct or stack axis; locate the probe tip at a single
preselected point of average velocity facing away from the direction of gas
flow. For stacks having a negative static pressure, seal the sample port
sufficiently to prevent air in-leakage around the probe. Set the probe
temperature controller to 129 ˚C (265 ˚F) and the filter temperature controller
to 121˚C (250 ˚F). Allow the probe and filter to heat for about 30 minutes
before purging the sample train.
8.2.2 Close the
sample valve, open the purge valve, and start the vacuum pump. Set the flow
rate between 60 and 100 cm3/min (0.13 and 0.21 ft3/hr), and purge the train with stack gas for at least 10 minutes.
8.2.3 When the
temperatures at the exit ends of the probe and filter are within the
corresponding specified ranges, check the dry ice level around the condensate
trap, and add dry ice if necessary. Record the clock time. To begin sampling,
close the purge valve and stop the pump. Open the sample valve and the sample
tank valve. Using the flow control valve, set the flow through the sample train
to the proper rate. Adjust the flow rate as necessary to maintain a constant
rate (±10 percent) throughout the duration of the sampling period. Record the
sample tank vacuum and flow meter setting at 5-minute intervals. (See Figure
25-8.) Select a total sample time greater than or equal to the minimum sampling
time specified in the applicable subpart of the regulations; end the sampling
when this time period is reached or when a constant flow rate can no longer be
maintained because of reduced sample tank vacuum.
NOTE: If sampling had to be stopped before obtaining
the minimum sampling time (specified in the applicable subpart) because a
constant flow rate could not be maintained, proceed as follows: After closing
the sample tank valve, remove the used sample tank from the sampling train
(without disconnecting other portions of the sampling train). Take another
evacuated and leak-checked sample tank, measure and record the tank vacuum, and
attach the new tank to the sampling train. After the new tank is attached to
the sample train, proceed with the sampling until the required minimum sampling
time has been exceeded.
8.3 Sample Recovery.
After sampling is
completed, close the flow control valve, and record the final tank vacuum; then
record the tank temperature and barometric pressure. Close the sample tank
valve, and disconnect the sample tank from the sample system. Disconnect the
condensate trap at the inlet to the rate meter, and tightly seal both ends of
the condensate trap. Do not include the probe from the stack to the filter as
part of the condensate sample.
8.4 Sample Storage and Transport.
Keep the trap packed
in dry ice until the samples are returned to the laboratory for analysis.
Ensure that run numbers are identified on the condensate trap and the sample
tank(s).
9.0 Quality Control.
10.0 Calibration and Standardization.
NOTE: Maintain a record of performance of each item.
10.1 Initial Performance Checks.
10.1.1 Condensate Recovery Apparatus.
Perform these tests
before the system is first placed in operation, after any shutdown of 6 months
or more, and after any major modification of the system, or at the frequency
recommended by the manufacturer.
10.1.1.1 Carrier Gas
and Auxiliary O2 Blank Check. Analyze each new tank of carrier
gas or auxiliary O2
with the NMO analyzer to check for
contamination. Treat the gas cylinders as non-condensible gas samples, and
analyze according to the procedure in Section 11.2.3. Add together any measured
CH4, CO, CO2, or NMO.
The total concentration must be less than 5 ppm.
10.1.1.2 Oxidation
Catalyst Efficiency Check.
10.1.1.2.1 With a
clean condensate trap installed in the recovery system or a 1/8" stainless
steel connector tube, replace the carrier gas cylinder with the high level
methane standard gas cylinder (Section 7.4.1). Set the four-port valve to the
recovery position, and attach an ICV to the recovery system. With the sample
recovery valve in vent position and the flow-control and ICV valves fully open,
evacuate the manometer or gauge, the connecting tubing, and the ICV to 10 mm Hg
absolute pressure. Close the flow-control and vacuum pump valves.
10.1.1.2.2 After the
NDIR response has stabilized, switch the sample recovery valve from vent to
collect. When the manometer or pressure gauge begins to register a slight
positive pressure, open the flow-control valve. Keep the flow adjusted such
that the pressure in the system is maintained within 10 percent of atmospheric
pressure. Continue collecting the sample in a normal manner until the ICV is
filled to a nominal gauge pressure of 300 mm Hg. Close the ICV valve, and
remove the ICV from the system. Place the sample recovery valve in the vent
position, and return the recovery system to its normal carrier gas and normal
operating conditions. Analyze the ICV for CO2 using the
NMO analyzer; the catalyst efficiency is acceptable if the CO2 concentration is within 2 percent of the methane standard
concentration.
10.1.1.3 System
Performance Check. Construct a liquid sample injection unit similar in design
to the unit shown in Figure 25-7. Insert this unit into the condensate recovery
and conditioning system in place of a condensate trap, and set the carrier gas
and auxiliary O2 flow rates to normal operating levels. Attach an
evacuated ICV to the system, and switch from system vent to collect. With the
carrier gas routed through the injection unit and the oxidation catalyst,
inject a liquid sample (see Sections 10.1.1.3.1 to 10.1.1.3.4) into the
injection port. Operate the trap recovery system as described in Section
11.1.3. Measure the final ICV pressure, and then analyze the vessel to determine
the CO2 concentration. For each injection, calculate the
percent recovery according to Section 12.7. Calculate the relative standard
deviation for each set of triplicate injections according to Section 12.8. The
performance test is acceptable if the average percent recovery is 100 ± 5
percent and the relative standard deviation is less than 2 percent for each set
of triplicate injections.
10.1.1.3.1 50 µl
hexane.
10.1.1.3.2 10 µl
hexane.
10.1.1.3.3 50 µl
decane.
10.1.1.3.4 10 µl
decane.
10.1.2 NMO Analyzer.
Perform these tests
before the system is first placed in operation, after any shutdown longer than
6 months, and after any major modification of the system.
10.1.2.1 Oxidation
Catalyst Efficiency Check. Turn off or bypass the NMO analyzer reduction
catalyst. Make triplicate injections of the high level methane standard
(Section 7.4.1). The oxidation catalyst operation is acceptable if the FID
response is less than 1 percent of the injected methane concentration.
10.1.2.2 Reduction
Catalyst Efficiency Check. With the oxidation catalyst unheated or bypassed and
the heated reduction catalyst bypassed, make triplicate injections of the
high-level methane standard (Section 7.4.1). Repeat this procedure with both
catalysts operative. The reduction catalyst operation is acceptable if the
responses under both conditions agree within 5 percent of their average.
10.1.2.3 NMO Analyzer
Linearity Check Calibration. While operating both the oxidation and reduction
catalysts, conduct a linearity check of the analyzer using the propane
standards specified in Section 7.4.2. Make triplicate injections of each
calibration gas. For each gas (i.e.,
each set of triplicate injections), calculate the average response factor
(area/ppm C) for each gas, as well as and the relative standard deviation
(according to Section 12.8). Then calculate the overall mean of the response
factor values. The instrument linearity is acceptable if the average response
factor of each calibration gas is within 2.5 percent of the overall mean value
and if the relative standard deviation gas is less than 2 percent of the
overall mean value. Record the overall mean of the propane response factor
values as the NMO calibration response factor (RFNMO).
Repeat the linearity check using the CO2 standards
specified in Section 7.4.3. Make triplicate injections of each gas, and then
calculate the average response factor (area/ppm C) for each gas, as well as the
overall mean of the response factor values. Record the overall mean of the
response factor values as the CO2 calibration response
factor (RFCO2). The RFCO2 must be
within 10 percent of the RFNMO.
10.1.2.4 System
Performance Check. Check the column separation and overall performance of the
analyzer by making triplicate injections of the calibration gases listed in
Section 7.4.4. The analyzer performance is acceptable if the measured NMO value
for each gas (average of triplicate injections) is within 5 percent of the
expected value.
10.2 NMO Analyzer Daily Calibration.
The following calibration
procedures shall be performed before and immediately after the analysis of each
set of samples, or on a daily basis, whichever is more stringent:
10.2.1 CO2 Response Factor.
Inject triplicate
samples of the high level CO2 calibration gas
(Section 7.4.3), and calculate the average response factor. The system
operation is adequate if the calculated response factor is within 5 percent of
the RFCO2 calculated during the initial performance test
(Section 10.1.2.3). Use the daily response factor (DRFCO2) for analyzer calibration and the calculation
of measured CO2 concentrations in the ICV samples.
10.2.2 NMO Response Factors.
Inject triplicate
samples of the mixed propane calibration cylinder gas (Section 7.4.4.1), and
calculate the average NMO response factor. The system operation is adequate if
the calculated response factor is within 10 percent of the RFNMO calculated during the initial performance test
(Section 10.1.2.4). Use the daily response factor (DRFNMO) for analyzer calibration and calculation of
NMO concentrations in the sample tanks.
10.3 Sample Tank and ICV Volume.
The volume of the gas
sampling tanks used must be determined. Determine the tank and ICV volumes by
weighing them empty and then filled with deionized distilled water; weigh to
the nearest 5 g, and record the results. Alternatively, measure the volume of
water used to fill them to the nearest 5 ml.
11.0 Analytical Procedure.
11.1 Condensate Recovery.
See Figure 25-9. Set
the carrier gas flow rate, and heat the catalyst to its operating temperature
to condition the apparatus.
11.1.1 Daily
Performance Checks. Each day before analyzing any samples, perform the
following tests:
11.1.1.1 Leak-Check.
With the carrier gas inlets and the sample recovery valve closed, install a clean
condensate trap in the system, and evacuate the system to 10 mm Hg absolute
pressure or less. Monitor the system pressure for 10 minutes. The system is
acceptable if the pressure change is less than 2 mm Hg.
11.1.1.2 System
Background Test. Adjust the carrier gas and auxiliary oxygen flow rate to their
normal values of 100 cc/min and 150 cc/min, respectively, with the sample
recovery valve in vent position. Using a 10-ml syringe, withdraw a sample from
the system effluent through the syringe port. Inject this sample into the NMO
analyzer, and measure the CO2 content. The system
background is acceptable if the CO2 concentration
is less than 10 ppm.
11.1.1.3 Oxidation
Catalyst Efficiency Check. Conduct a catalyst efficiency test as specified in
Section 10.1.1.2. If the criterion of this test cannot be met, make the
necessary repairs to the system before proceeding.
11.1.2 Condensate
Trap CO2 Purge and Sample Tank Pressurization.
11.1.2.1 After
sampling is completed, the condensate trap will contain condensed water and
organics and a small volume of sampled gas. This gas from the stack may contain
a significant amount of CO2
which must be removed from the
condensate trap before the sample is recovered. This is accomplished by purging
the condensate trap with zero air and collecting the purged gas in the original
sample tank.
11.1.2.2 Begin with
the sample tank and condensate trap from the test run to be analyzed. Set the
four-port valve of the condensate recovery system in the CO2 purge position as shown in Figure 25-9. With the sample tank valve
closed, attach the sample tank to the sample recovery system. With the sample
recovery valve in the vent position and the flow control valve fully open,
evacuate the manometer or pressure gauge to the vacuum of the sample tank.
Next, close the vacuum pump valve, open the sample tank valve, and record the
tank pressure.
11.1.2.3 Attach the
dry ice-cooled condensate trap to the recovery system, and initiate the purge
by switching the sample recovery valve from vent to collect position. Adjust
the flow control valve to maintain atmospheric pressure in the recovery system.
Continue the purge until the CO2 concentration of the
trap effluent is less than 5 ppm. CO2 concentration
in the trap effluent should be measured by extracting syringe samples from the
recovery system and analyzing the samples with the NMO analyzer. This procedure
should be used only after the NDIR response has reached a minimum level. Using
a 10-ml syringe, extract a sample from the syringe port prior to the NDIR, and
inject this sample into the NMO analyzer.
11.1.2.4 After the
completion of the CO2
purge, use the carrier gas bypass
valve to pressurize the sample tank to approximately 1,060 mm Hg absolute
pressure with zero
air.
11.1.3 Recovery of
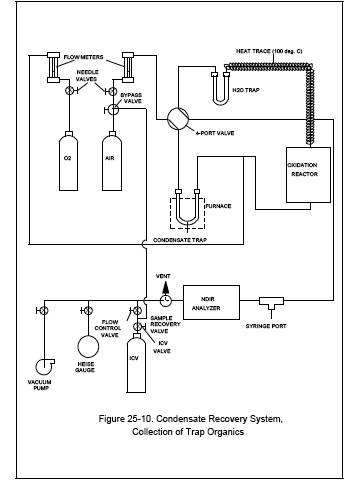
the Condensate Trap Sample (See Figure 25-10).
11.1.3.1 Attach the
ICV to the sample recovery system. With the sample recovery valve in a closed
position, between vent and collect, and the flow control and ICV valves fully
open, evacuate the manometer or gauge, the connecting tubing, and the ICV to 10
mm Hg absolute pressure. Close the flow-control and vacuum pump valves.
11.1.3.2 Begin
auxiliary oxygen flow to the oxidation catalyst at a rate of 150 cc/min, then
switch the four-way valve to the trap recovery position and the sample recovery
valve to collect position. The system should now be set up to operate as
indicated in Figure 25-10. After the manometer or pressure gauge begins to
register a slight positive pressure, open the flow control valve. Adjust the
flow-control valve to maintain atmospheric pressure in the system within 10
percent.
11.1.3.3 Remove the
condensate trap from the dry ice, and allow it to warm to ambient temperature
while monitoring the NDIR response. If, after 5 minutes, the CO2 concentration of the catalyst effluent is below 10,000 ppm,
discontinue the auxiliary oxygen flow to the oxidation catalyst. Begin heating
the trap by placing it in a furnace preheated to 200 ˚C (390 ˚F). Once heating
has begun, carefully monitor the NDIR response to ensure that the catalyst
effluent concentration does not exceed 50,000 ppm. Whenever the CO2 concentration exceeds 50,000 ppm, supply auxiliary oxygen to the
catalyst at the rate of 150 cc/min. Begin heating the tubing that connected the
heated sample box to the condensate trap only after the CO2 concentration falls below 10,000 ppm. This tubing may be heated in
the same oven as the condensate trap or with an auxiliary heat source such as a
heat gun. Heating temperature must not exceed 200 ˚C (390 ˚F). If a heat gun is
used, heat the tubing slowly along its entire length from the upstream end to
the downstream end, and repeat the pattern for a total of three times. Continue
the recovery until the CO2
concentration drops to less than 10
ppm as determined by syringe injection as described under the condensate trap
CO2 purge procedure (Section 11.1.2).
11.1.3.4 After the
sample recovery is completed, use the carrier gas bypass valve to pressurize
the ICV to approximately 1060 mm Hg absolute pressure with zero air.
11.2 Analysis.
Once the initial
performance test of the NMO analyzer has been successfully completed (see
Section 10.1.2) and the daily CO2 and NMO response
factors have been determined (see Section 10.2), proceed with sample analysis
as follows:
11.2.1 Operating
Conditions. The carrier gas flow rate is 29.5 cc/min He and 2.2 cc/min O2. The column oven is heated to 85 ˚C (185 ˚F). The order of elution
for the sample from the column is CO, CH4, CO2, and NMO.
11.2.2 Analysis of
Recovered Condensate Sample. Purge the sample loop with sample, and then inject
the sample. Under the specified operating conditions, the CO2 in the sample will elute in approximately 100 seconds. As soon as
the detector response returns to baseline following the CO2 peak, switch the carrier gas flow to back-flush, and raise the
column oven temperature to 195 ˚C (380 ˚F) as rapidly as possible. A rate of 30
˚C/min (90 ˚F) has been shown to be adequate. Record the value obtained for the
condensible organic material (Ccm) measured as CO2 and any measured NMO. Return the column oven temperature to 85 ˚C
(185 ˚F) in preparation for the next analysis. Analyze each sample in
triplicate, and report the average Ccm.
11.2.3 Analysis of
Sample Tank. Perform the analysis as described in Section 11.2.2, but record
only the value measured for NMO (Ctm).
11.3 Audit Sample Analysis.
11.3.1 When the
method is used to analyze samples to demonstrate compliance with a source
emission regulation, an audit sample, if available, must be analyzed.
11.3.2 Concurrently
analyze the audit sample and the compliance samples in the same manner to
evaluate the technique of the analyst and the standards preparation.
11.3.3 The same
analyst, analytical reagents, and analytical system must be used for the
compliance samples and the audit sample. If this condition is met, duplicate
auditing of subsequent compliance analyses for the same enforcement agency
within a 30-day period is waived. An audit sample set may not be used to
validate different sets of compliance samples under the jurisdiction of
separate enforcement agencies, unless prior arrangements have been made with
both enforcement agencies.
11.4 Audit Sample Results.
11.4.1 Calculate the
audit sample concentrations and submit results using the instructions provided
with the audit samples.
11.4.2 Report the
results of the audit samples and the compliance determination samples along
with their identification numbers, and the analyst's name to the responsible
enforcement authority. Include this information with reports of any subsequent
compliance analyses for the same enforcement authority during the 30-day
period.
11.4.3 The
concentrations of the audit samples obtained by the analyst must agree within
20 percent of the actual concentration. If the 20-percent specification is not
met, reanalyze the compliance and audit samples, and include initial and
reanalysis values in the test report.
11.4.4 Failure to
meet the 20-percent specification may require retests until the audit problems
are resolved. However, if the audit results do not affect the compliance or
noncompliance status of the affected facility, the Administrator may waive the
reanalysis requirement, further audits, or retests and accept the results of
the compliance test. While steps are being taken to resolve audit analysis
problems, the Administrator may also choose to use the data to determine the
compliance or noncompliance status of the affected facility.
12.0 Data Analysis and Calculations.
Carry out the calculations,
retaining at least one extra significant figure beyond that of the acquired
data. Round off figures after final calculations. All equations are written
using absolute pressure; absolute pressures are determined by adding the
measured barometric pressure to the measured gauge or manometer pressure.
12.1 Nomenclature.
C = TGNMO
concentration of the effluent, ppm C equivalent.
Cc = Calculated condensible organic (condensate trap) concentration of
the effluent, ppm C equivalent.
Ccm = Measured concentration (NMO analyzer) for the
condensate trap ICV, ppm CO2.
Ct = Calculated noncondensible organic concentration (sample tank) of
the effluent, ppm C equivalent.
Ctm = Measured concentration (NMO analyzer) for the
sample tank, ppm NMO.
F = Sampling flow
rate, cc/min.
L = Volume of liquid
injected, µl.
M = Molecular weight
of the liquid injected, g/gmole.
Mc = TGNMO mass concentration of the effluent, mg C/dsm3.
N = Carbon number of
the liquid compound injected
(N = 12 for decane, N
= 6 for hexane).
n = Number of data
points.
Pf = Final pressure of the intermediate collection vessel, mm Hg
absolute.
Pb = Barometric pressure, cm Hg.
Pti = Gas sample tank pressure before sampling, mm
Hg absolute.
Pt = Gas sample tank pressure after sampling, but before pressurizing,
mm Hg absolute.
Ptf = Final gas sample tank pressure after
pressurizing, mm Hg absolute.
q = Total number of
analyzer injections of intermediate collection vessel during analysis
(where k = injection
number, 1 ... q).
r = Total number of
analyzer injections of sample tank during analysis (where j = injection number,
1 ... r).
r = Density of liquid
injected, g/cc.
Tf = Final temperature of intermediate collection vessel, ˚K.
Tti = Sample tank temperature before sampling, ˚K.
Tt = Sample tank temperature at completion of sampling, ˚K.
Ttf = Sample tank temperature after pressurizing,
˚K.
V = Sample tank
volume, m3.
Vt = Sample train volume, cc.
Vv = Intermediate collection vessel volume, m3.
Vs = Gas volume sampled, dsm3.
xi = Individual measurements.
x = Mean value.
•P = Allowable
pressure change, cm Hg.
• = Leak-check
period, min.
12.2 Allowable
Pressure Change. For the pretest leak-check, calculate the allowable pressure
change using Equation 25-1:

12.3 Sample Volume. For
each test run, calculate the gas volume sampled using Equation 25-2:
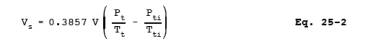
12.4 Non-condensible
Organics. For each sample tank, determine the concentration of non-methane
organics (ppm C) using Equation 25-3:
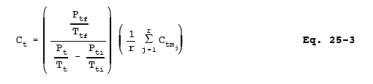
12.5 Condensible
Organics. For each condensate trap determine the concentration of organics (ppm
C) using Equation 25-4:
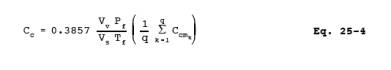
12.6 TGNMO Mass
Concentration. Determine the TGNMO mass concentration as carbon for each test
run, using Equation 25-5:
![]()
12.7 Percent Recovery.
Calculate the percent recovery for the liquid injections to the condensate
recovery and conditioning system using Equation 25-6:
![]()
where K = 1.604
(˚K)(g-mole)(%)/(mm Hg)(ml)(m3)(ppm).
12.8 Relative
Standard Deviation. Use Equation 25-7 to calculate the relative standard
deviation (RSD) of percent recovery and analyzer linearity.
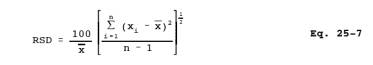
13.0 Method Performance.
13.1 Range. The
minimum detectable limit of the method has been determined to be 50 parts per
million by volume (ppm). No upper limit has been established.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
1. Salo, A.E., S.
Witz, and R.D. MacPhee. Determination of Solvent Vapor Concentrations by Total Combustion
Analysis: A Comparison of Infrared with Flame Ionization Detectors. Paper No.
75-33.2. (Presented at the 68th Annual Meeting of the Air Pollution Control
Association. Boston, MA. June 15-20, 1975.) 14 p.
2. Salo, A.E., W.L. Oaks,
and R.D. MacPhee. Measuring the Organic Carbon Content of Source Emissions for
Air Pollution Control. Paper No. 74-190. (Presented at the 67th Annual Meeting
of the Air Pollution Control Association. Denver, CO. June 9-13, 1974.) 25 p.
17.0 Tables, Diagrams, Flowcharts, and Validation Data.
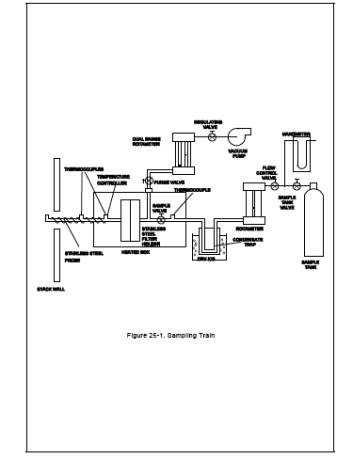
Figure 25-2. Out-of-stack Filter Box
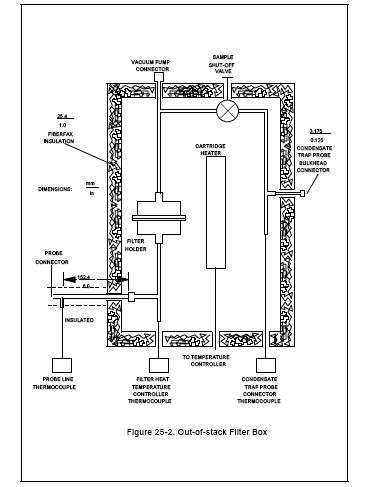
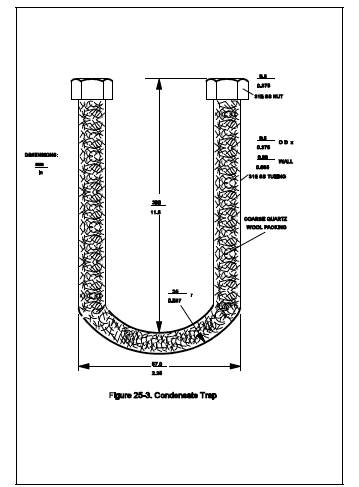
Figure 25-4.
Condensate Recovery System
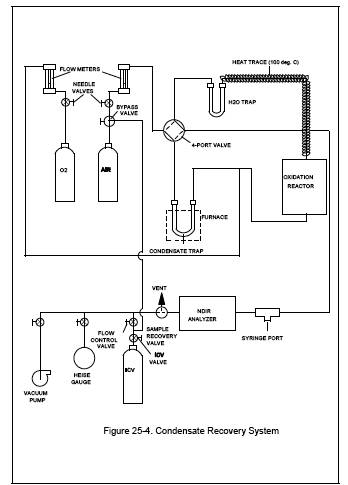
Figure 25-5. Simplified
Schematic of Nonmethane Organic (NMO) Analyzer
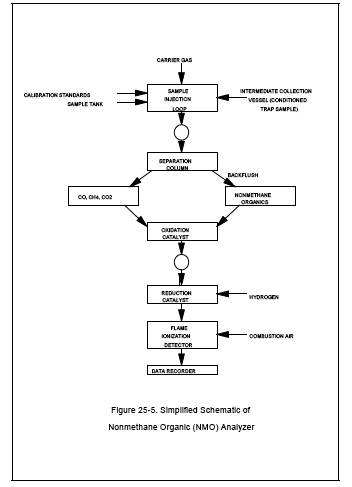
Figure 25-6.
Nonmethane Organic Analyzer (NMO)
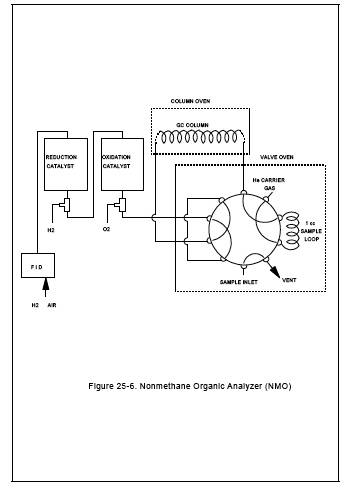
Figure 25-7.
Liquid Sample Injection Unit
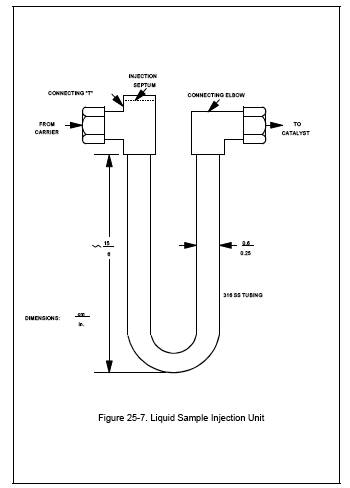
Figure 25-8.
Example Field Data Form

Figure 25-9. Condensate
Recovery System, CO2 Purge
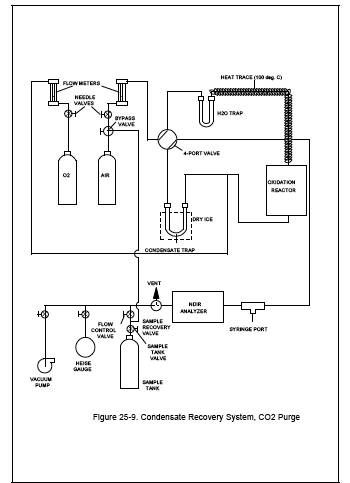
Figure 25-10.
Condensate Recovery System,